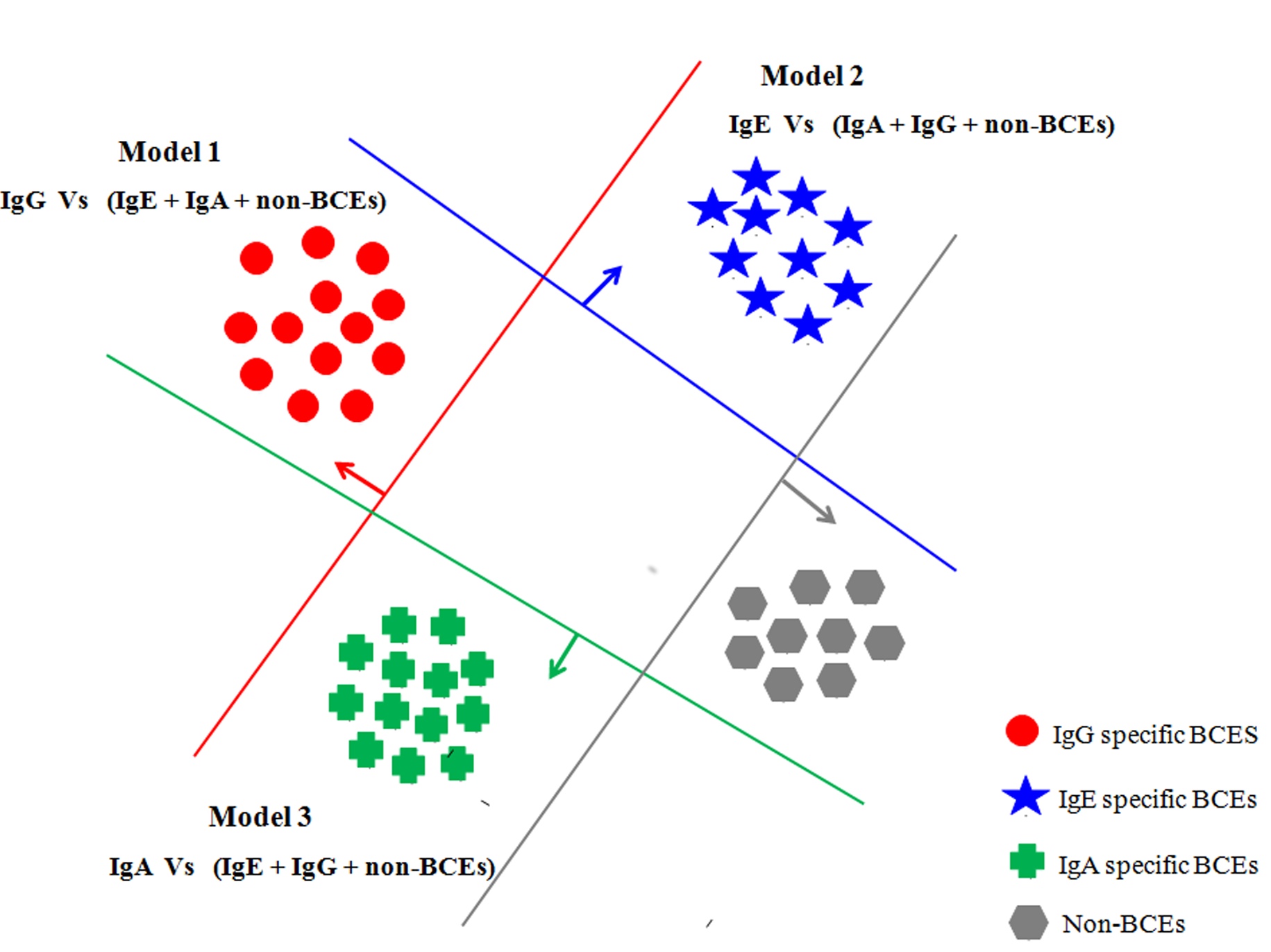
|
1. Amino Acid Composition: Amino Acid Composition is the fraction of each amino acid present in a peptide. There are 20 vectors generated in which one corresponds to one amino acid and these vectors used for as SVM input.
2. Dipeptide Composition: Dipeptide Composition is the fraction of each dipeptide like AA, AC, AD and so on. It provides compositional as well as local order each residue present in the peptide. It contains 20x20 (400) vectors.
3. Amino Acid Propensity: Amino Acid Propensity can be defined as the dipeptide composition multiplied by its frequency of occurrence in Bcipep and Swissprot databases. Here vector size remains 400.
4. Composition-transition -distribution: Each peptide sequence is mapped in to a string defined by three symbols. These symbols are resulted from grouping of all amino acids in to three groups, on the basis of certain physiochemical property. For every physicochemical property, we have string of 1,2 and 3 symbols, three feature given by composition three feature given by the percent frequency of i followed by j or j followed by i (transition) and three features are five features per symbol representing the fractions of the entire sequence where the first, 25, 50, 75, and 100% of the candidate symbol are contained in string (distribution). The final vector size becomes 108.
5. Physico-chemical Properties: Physico-chemical Properties of each amino acid like hydrophobicity, hydrohpilicity, charge, pI etc. has been used as input feature for the prediction. We obtained physico-chemical properties values of each amino acid form the webserver AAindex and used them to calculate physico-chemical properties of peptide by Perl programes.
5. Binary Profile: In binary profile of patterns, each amino acid is presented by a vector of dimension 20 as described below. Since the length of epitopes was 20, a pattern of window length 20 is represented by a vector of dimension (20 x 20) .

|
|