Algorithm
Dataset
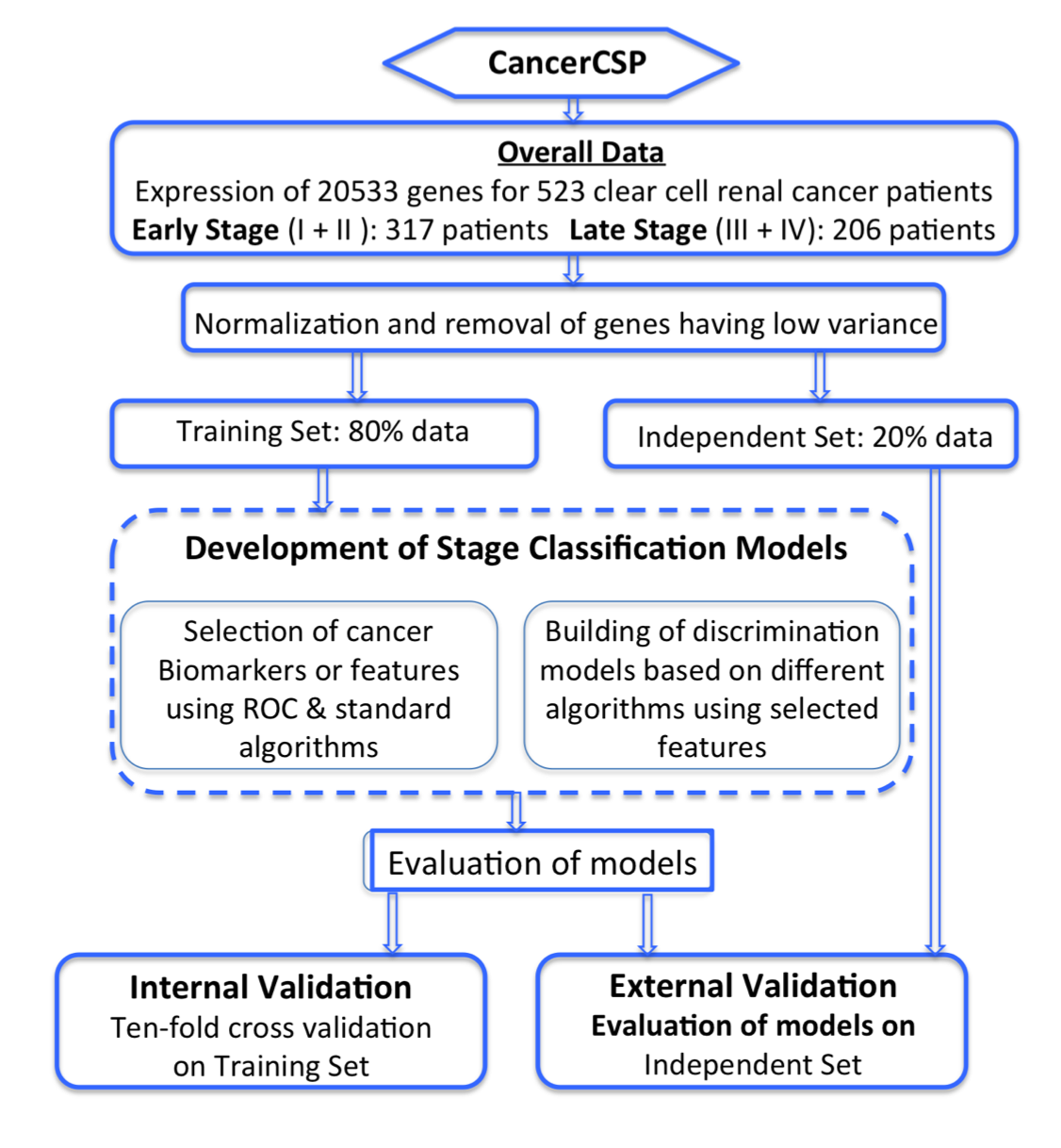
The Level 3 RNA-seq expression data for Kidney Renal clear cell carcinoma (KIRC) was obtained from TCGA data portal (https://tcgadata.nci.nih.gov/tcga/dataAccessMatrix.html). We selected Level 3 data for all types of data in RNA-seq version in February 2015. The data obtained consisted of raw count and RSEM values of gene quantification for 20,501 genes for different patients. RSEM was used as quantification values and only tumor samples with matched normal or unmatched normal were taken in this study. Corresponding clinical information for the patients was based on the BCR (Biospecimen Core Resource) IDs of the patients and was obtained from “clinical biotab” section of the data matrix. We organized the data consequently and made a complete matrix with 20,501 columns (genes) for a variable number of patients related to renal cancer. Each data point represents RSEM values (Li et al., 2010). We took 20,501 genes for which symbols are given and log2 transformed RSEM values were used as input features for classifier training. For binary classification we tagged stage I and stage II patients as early stage patients and stage III and stage IV patients as late stage patients. Further we divided the data in 80%-20% ratio after random shuffling of samples for training and blind dataset for validation purpose using in-house perl, python and R scripts.
Feature Selection
Feature selection is implemented using Waikato Environment for Knowledge Analysis (WEKA) (Ian H. Witten, 2000) using attribute evaluator named, ‘SymmetricalUncertAttributeSetEval’ with search method of ‘FCBFSearch’. The algorithm Fast Correlation Based Feature (FCBF) selection utilizes predominant correlation to identify relevant features in high dimensional datasets in very less time (Yu L, 2003). Next for classification models we implemented WEKA for classification and validation of models. These features were selected separately in 80% of the data for renal cancer.
Log and Probability Analysis
As the range of data is very high the log transformation of data is done to improve the interpretability of the data.
Analysis
Mean expression of selected features for renal cancer in early and late patients is calculated and compared to the gene expression of the patient in the form of bar graph.
Evaluation of Performance
Five-fold cross validation technique has been used. Four sets are used for training and remaining one in used for testing, in this way the process repeats five times. Evaluation of performance of different SVM modules has been done by calculating accuracy and Matthew's correlation coefficient (MCC).