About Hemolytic peptides
What are Hemolytic Peptides?
Hemolytic peptides are short chains of amino acids that can disrupt red blood cell (RBC) membranes, leading to cell rupture — a process known as hemolysis. These peptides are often found in nature, such as in snake venoms, antimicrobial peptides, or human immune proteins.
How is This Useful in Therapeutics?
Predicting and controlling hemolytic activity supports multiple therapeutic advancements:
- Antimicrobial Peptides (AMPs): Non-hemolytic AMPs combat drug-resistant bacteria without harming human cells.
- Cancer Therapy: Some hemolytic peptides selectively target cancer cells — modifying them improves safety.
- Drug Delivery: Hemolytic peptides can enhance drug delivery across membranes when tuned to target diseased cells specifically.
- Antiviral and Anti-fungal Peptides: Safer, modified peptides can fight viruses or fungi without harming human cells.
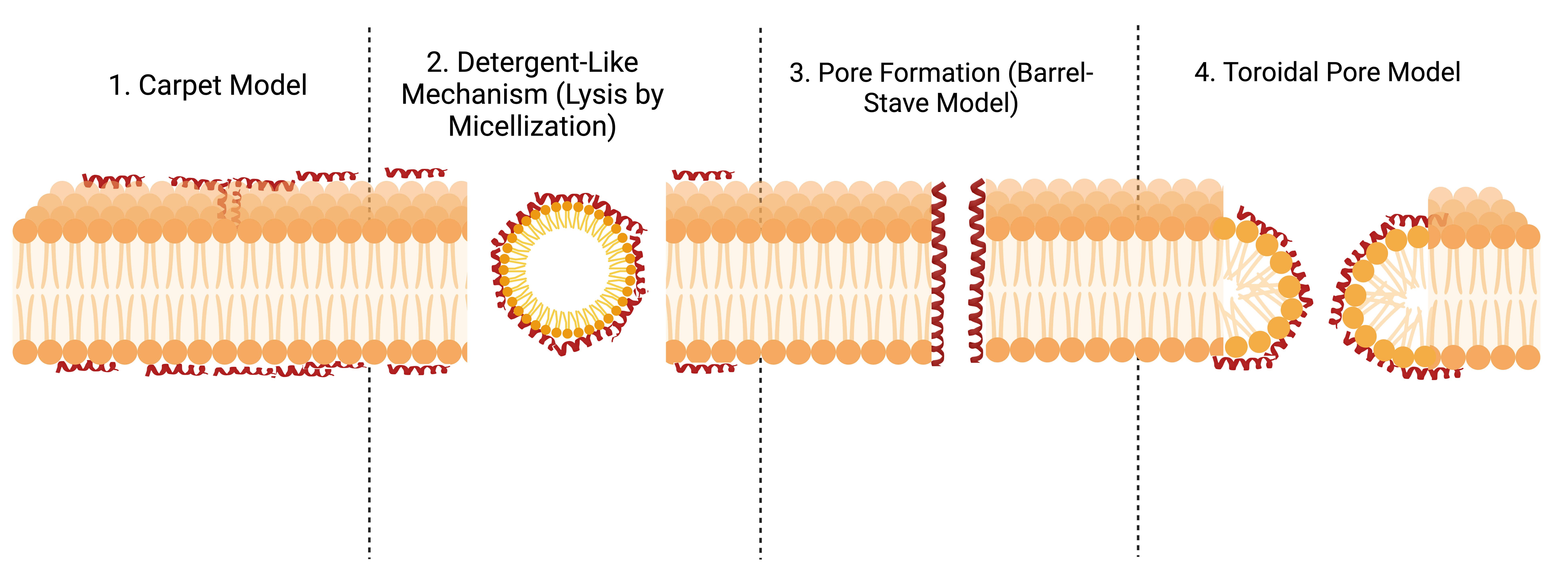
Hemolytic Peptides: Action Mechanisms
1. Carpet Model
Peptides cover the membrane surface like a carpet. When a critical concentration is reached, the membrane disintegrates.
Example: Cecropins (insects)
Analogy: Like detergent breaking down grease — the membrane dissolves.
3. Detergent-Like Mechanism (Lysis by Micellization)
Peptides integrate into the membrane, causing phospholipids to rearrange into tiny vesicles (micelles), destabilizing the membrane.
Example: LL-37 (human antimicrobial peptide)
Analogy: Like shaking an oil-water mix until droplets form, breaking the structure.
3. Pore Formation (Barrel-Stave Model)
Peptides align parallel to the cell membrane, then insert themselves perpendicularly to form a cylindrical pore. This allows ions and water to flow uncontrollably, leading to cell lysis.
Example: Melittin (bee venom)
Analogy: Like drilling a hole in a balloon — the contents spill out, causing collapse.
4. Toroidal Pore Model
Peptides bend the membrane itself, pulling the lipid heads inward to form a pore. The membrane and peptides intertwine in the structure.
Example: Magainins (amphibians)
Analogy: Like stretching a rubber band until it folds inward to form a hole.
Why This Matters for Therapeutics
Understanding these mechanisms helps design safer peptides:
- Pore-forming peptides: Can be modified to selectively target cancer or bacterial cells.
- Carpet-like peptides: Can be engineered to avoid damaging human cells.
- Toroidal model peptides: Provide insights into creating peptides that penetrate tough bacterial membranes while sparing human ones.
Search
This section provides information about how to perform searches.
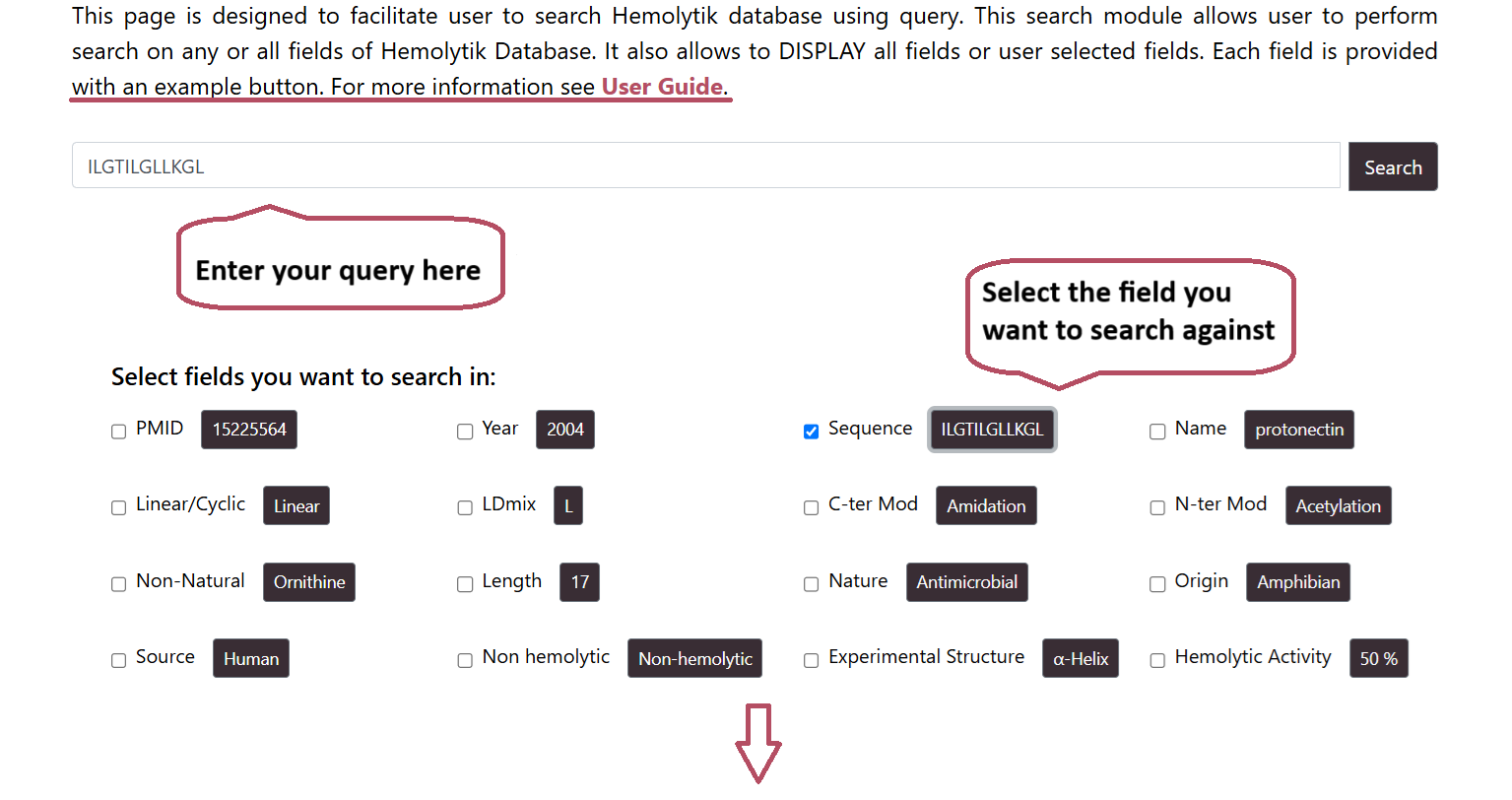
Basic Search
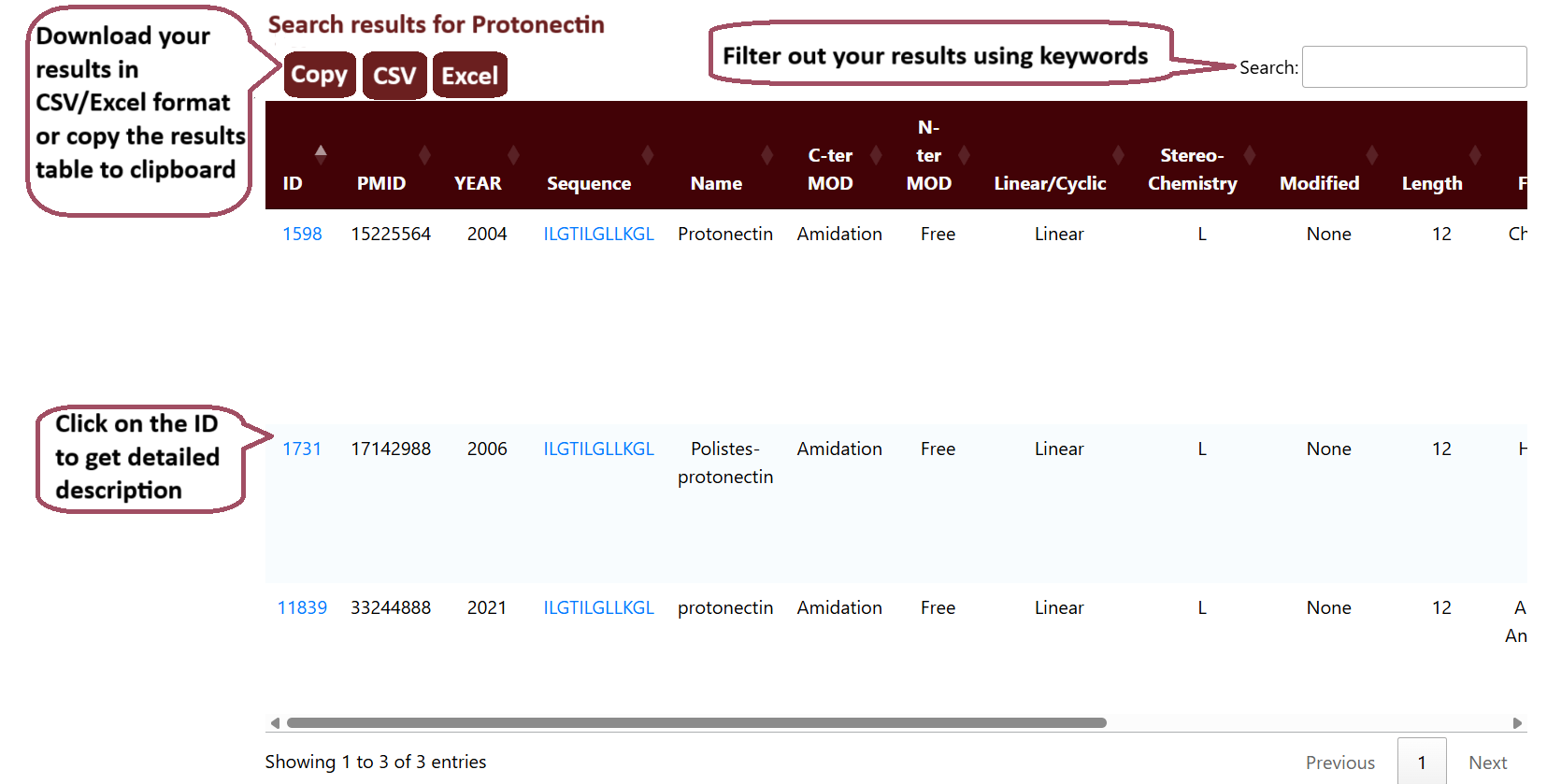
The user can input a search term in the search field and select the checkboxes to choose the specific fields they want to search.
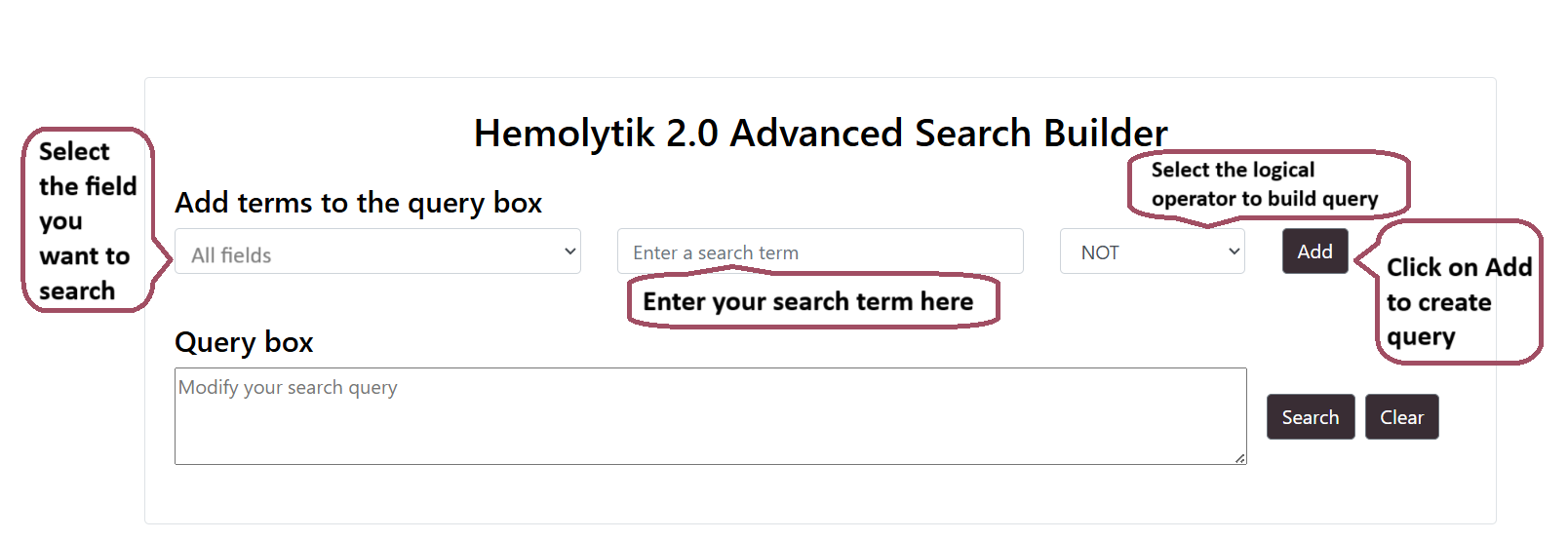
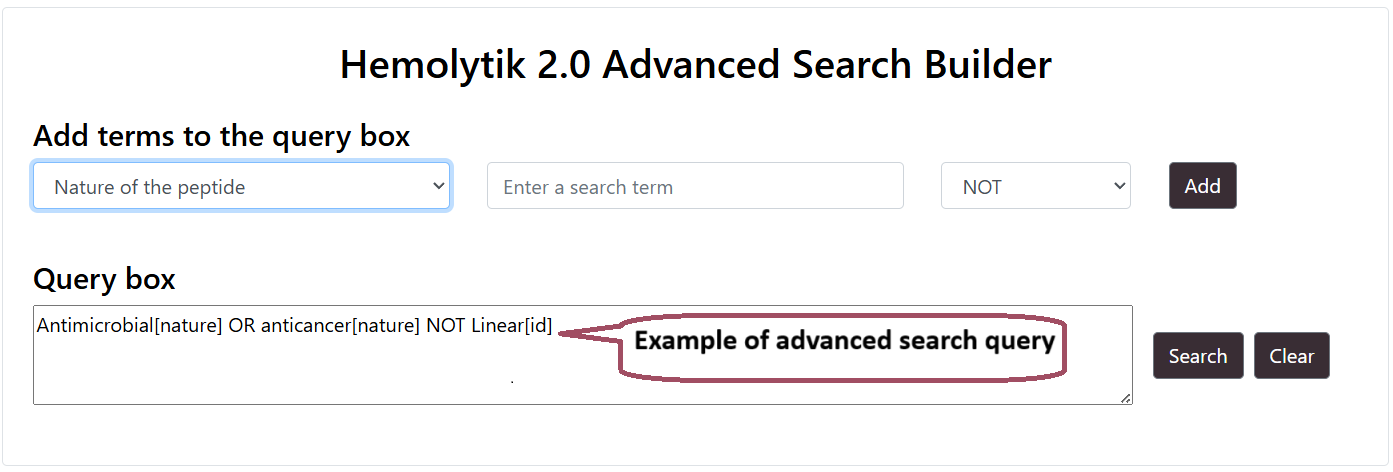
Advance Search
Users can perform advance search by selecting multiple fields using logical operators like AND/OR. Users can build the query by using the "Add" button.
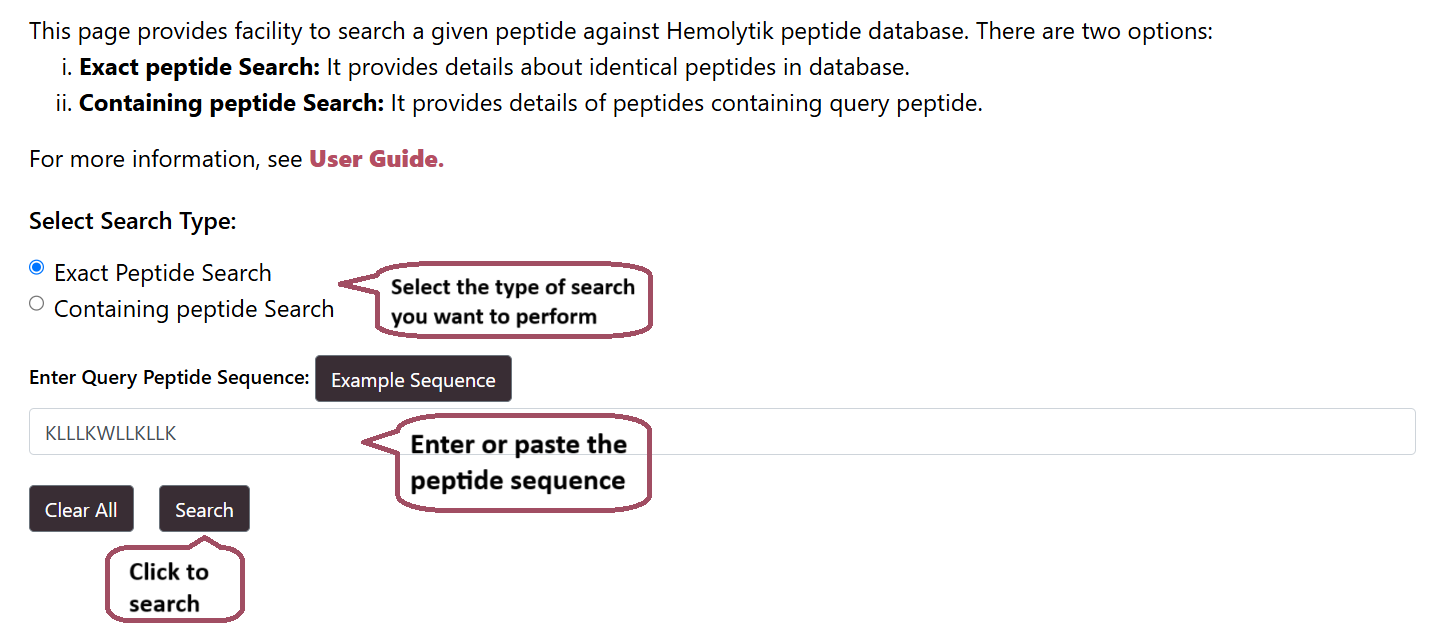
Peptide Search
Users can perform Peptide search to search for their query peptide sequence in the Hemolytik 2.0 Database. The module provides two search options:
- Identical Sequence Search: This option will search for identical peptide sequence in the database.
- Subsequence Search: This option will will search for peptides that contains a part of the query peptide.
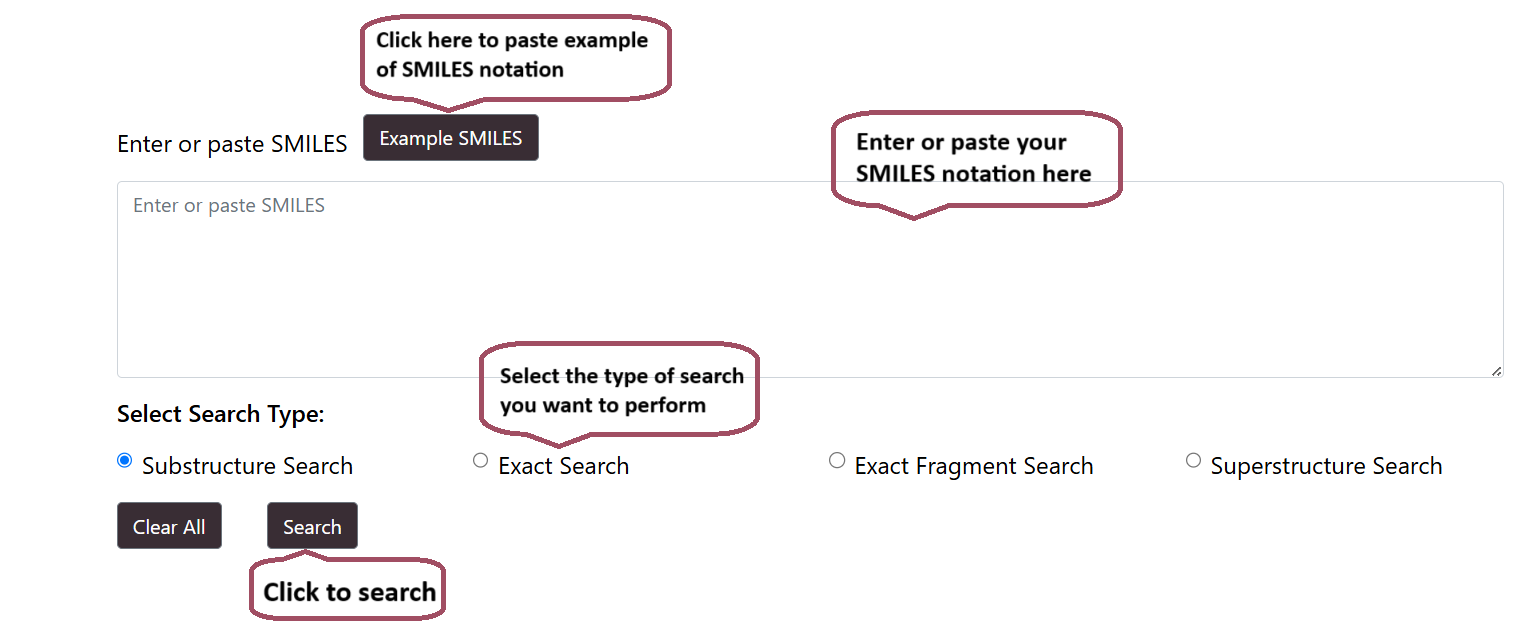
SMILES Search
Users can perform SMILES search to search for the query SMILES sequence in the Hemolytik 2.0 Database. The module provides four search options:
- Substructure Search
- Exact Search
- Exact Fragment Search
- Superstructure Search
MAP Format
What is MAP Format?
MAP (Modified Amino acid Peptide) format is a structured representation specifically designed for modified peptides, particularly those containing non-natural residues or chemical modifications that aren't covered by standard one-letter amino acid codes. This format ensures consistency in representing peptide sequences across different computational tools and databases.
Why is MAP Format Important?
In peptide-based therapeutics and bioinformatics, especially when working with hemolytic peptides, accurate representation of both natural and non-natural residues is crucial. MAP format provides a reliable way to encode these sequences for computational analysis, predictive modeling, and database storage.
Need to Convert Your Sequences?
If you have raw peptide sequences and want to convert them into MAP format, you can easily do so using the tools and guidelines provided in the MAP repository developed by Raghava's group. This resource includes conversion utilities, examples, and detailed documentation to help you get started.
Explore the MAP Repository: https://webs.iiitd.edu.in/raghava/maprepo/
Dive deeper into MAP format structure, guidelines for representing modified residues, and how to use MAP files in peptide analysis workflows.
Browse
This section provides information about browsing the Hemolytik 2.0 entries by different ways.
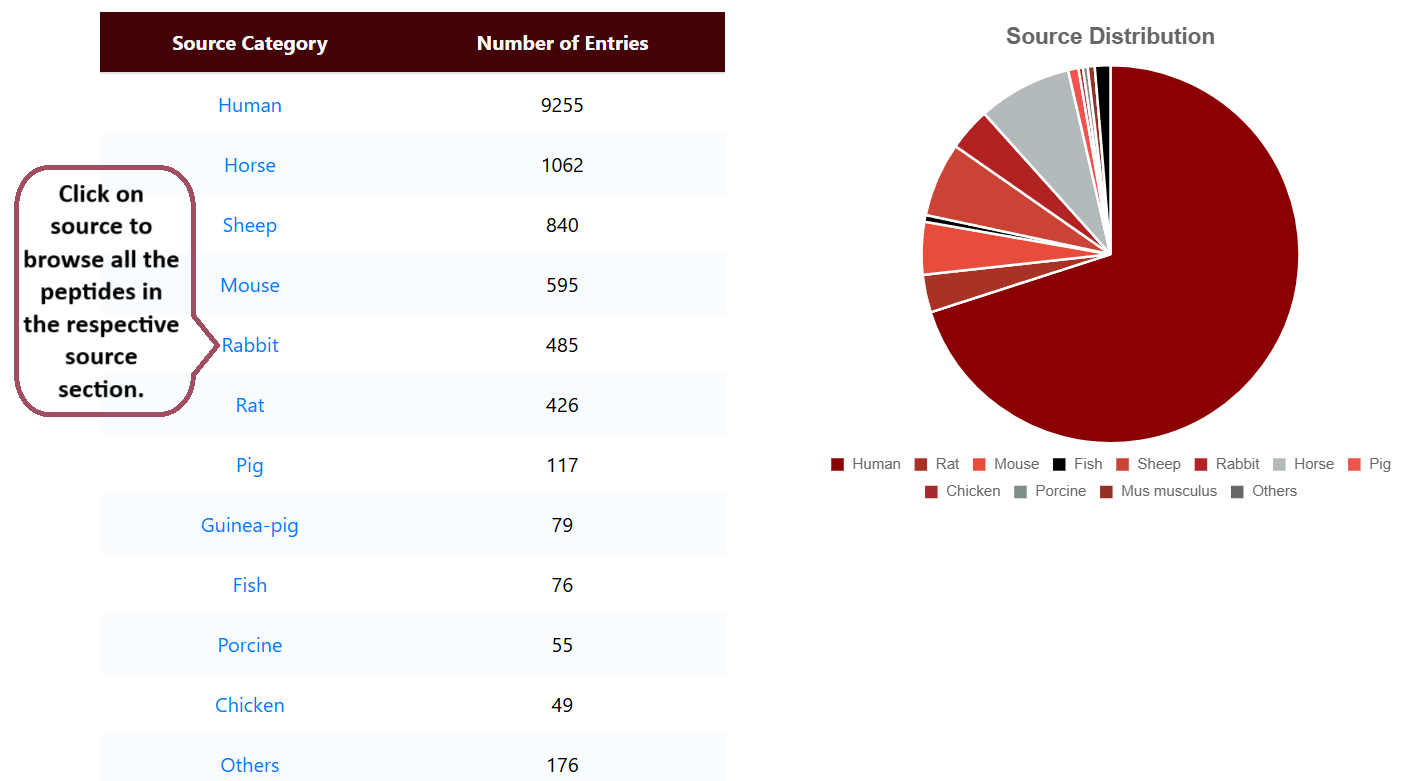
Browse by Source
Users can browse different sources of peptides or proteins.
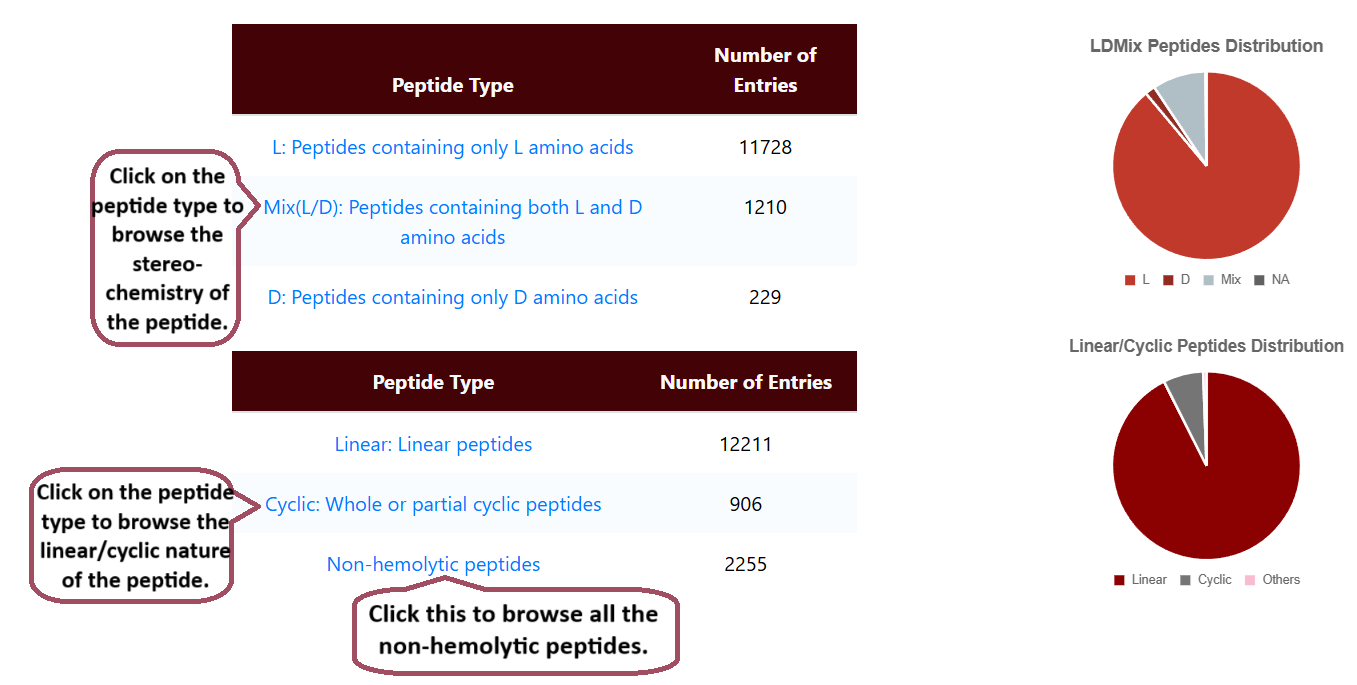
Browse by Function
Users can browse the function of hemolytic peptides. It lists the number of entries of linear and cyclic peptides, the stereochemistry of peptides, and the number of non-hemolytic peptides.
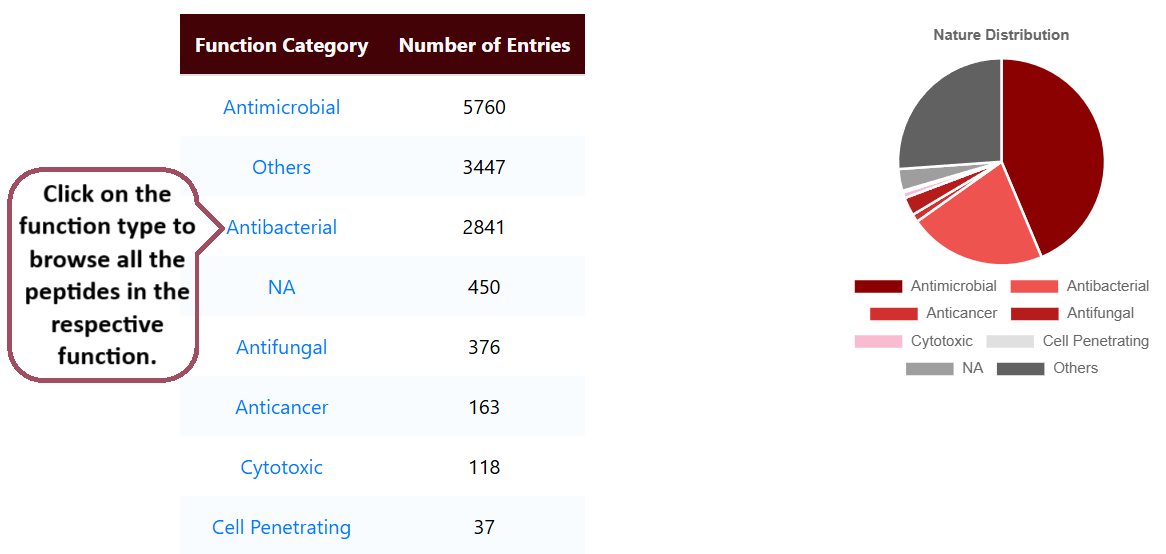
Browse by Nature
Users can browse the unique hemolytic peptides by their nature. It lists the number of entries for different nature of peptides.
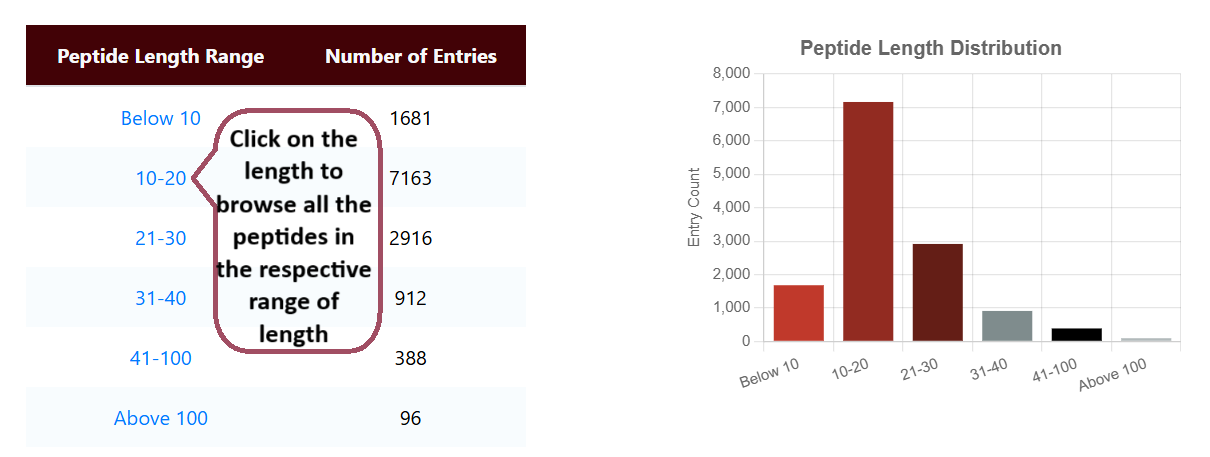
Browse by Length
Users can browse hemolytic peptides in Hemolytik 2.0 based on their length. Sequences up to 40 amino acids are classified as peptides, while those exceeding 40 amino acids are classified as proteins.
Peptide Card
The peptide card contains all the information about a given peptide or protein. The peptide card contains the Basic Information such as PMID, origin, Sequence, Length etc., Information about Modifications such as Chemical Modifications, C-Ter Modifications, N-Ter Modifications, Experimental Data such as Activity, Assay, Cell Line etc., Literature Information such as Title of the paper, DOI, Journal, Abstract etc., Secondary Infromation (DSSP and SMILES) and External Links to PDB, SwissProt or TrEMBL.
The peptide and protein structures present in Hemolytik 2.0 includes both structures from PDB (experimental) and Predicted Structures. The structures also contains some associated data.
- DSSP States
- SMILES
Peptide Card of a Entry in Hemolytik 2.0
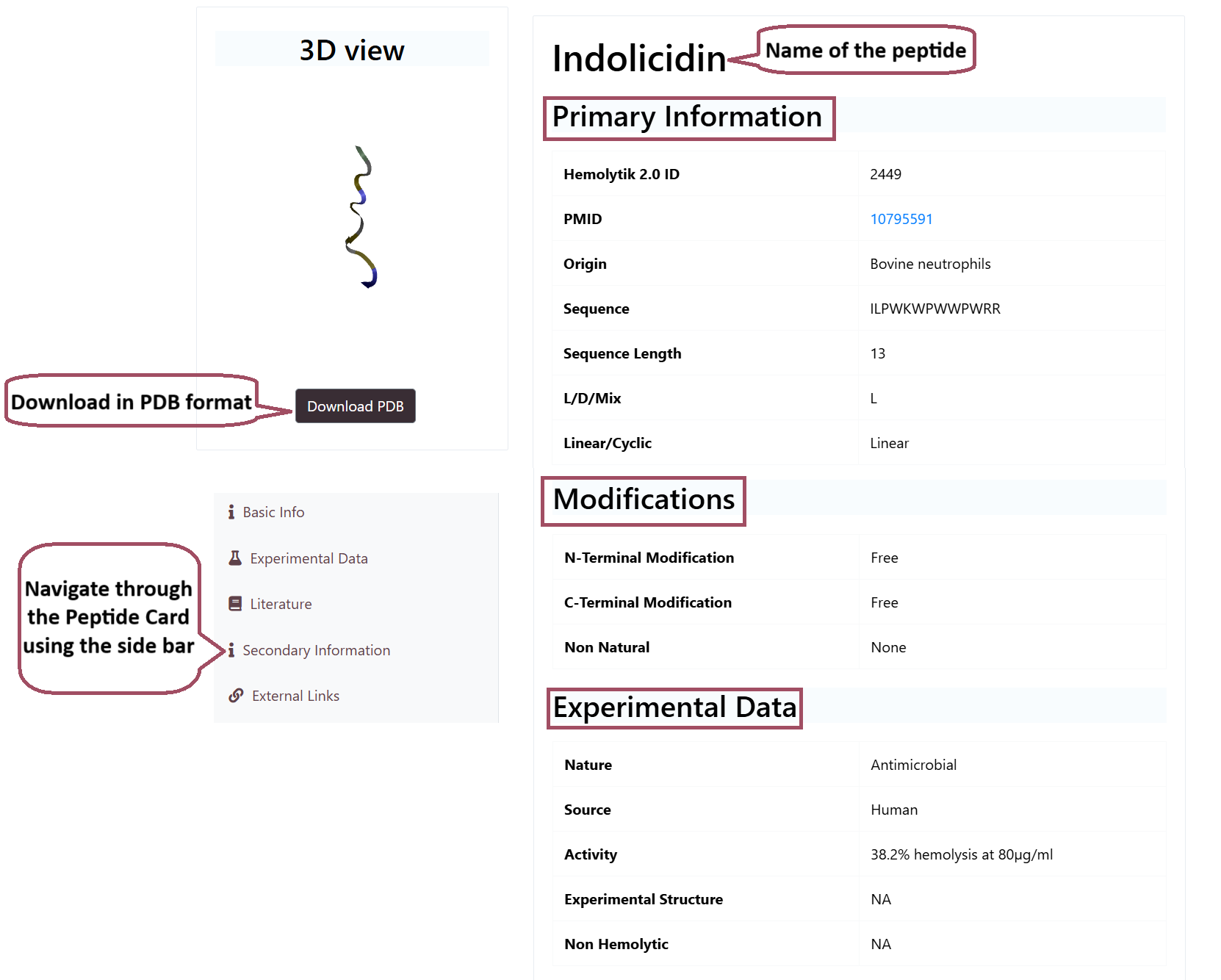
3D Structure
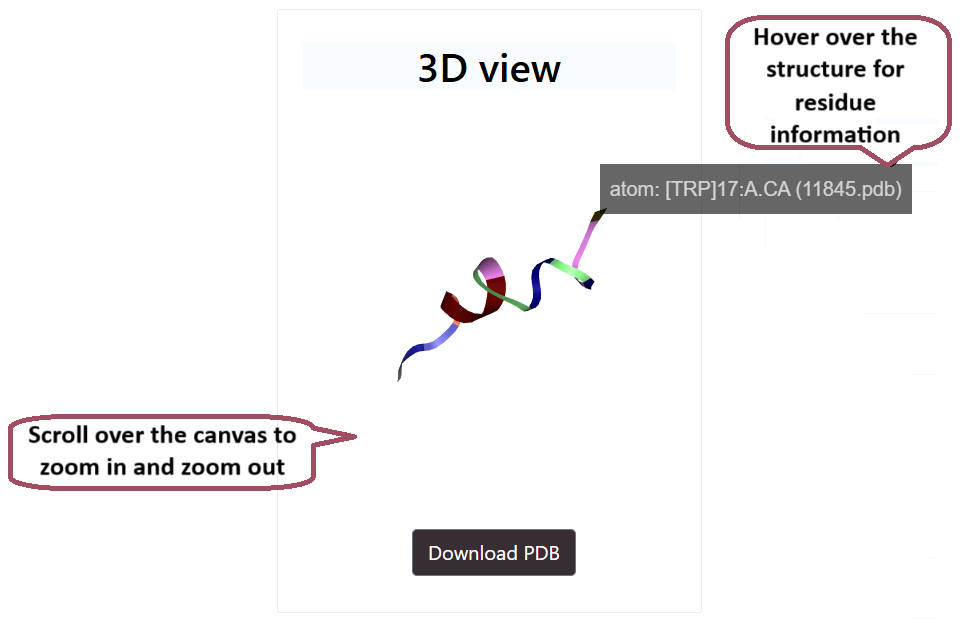
Tools
This section provides information about the tools available in Hemolytik 2.0.
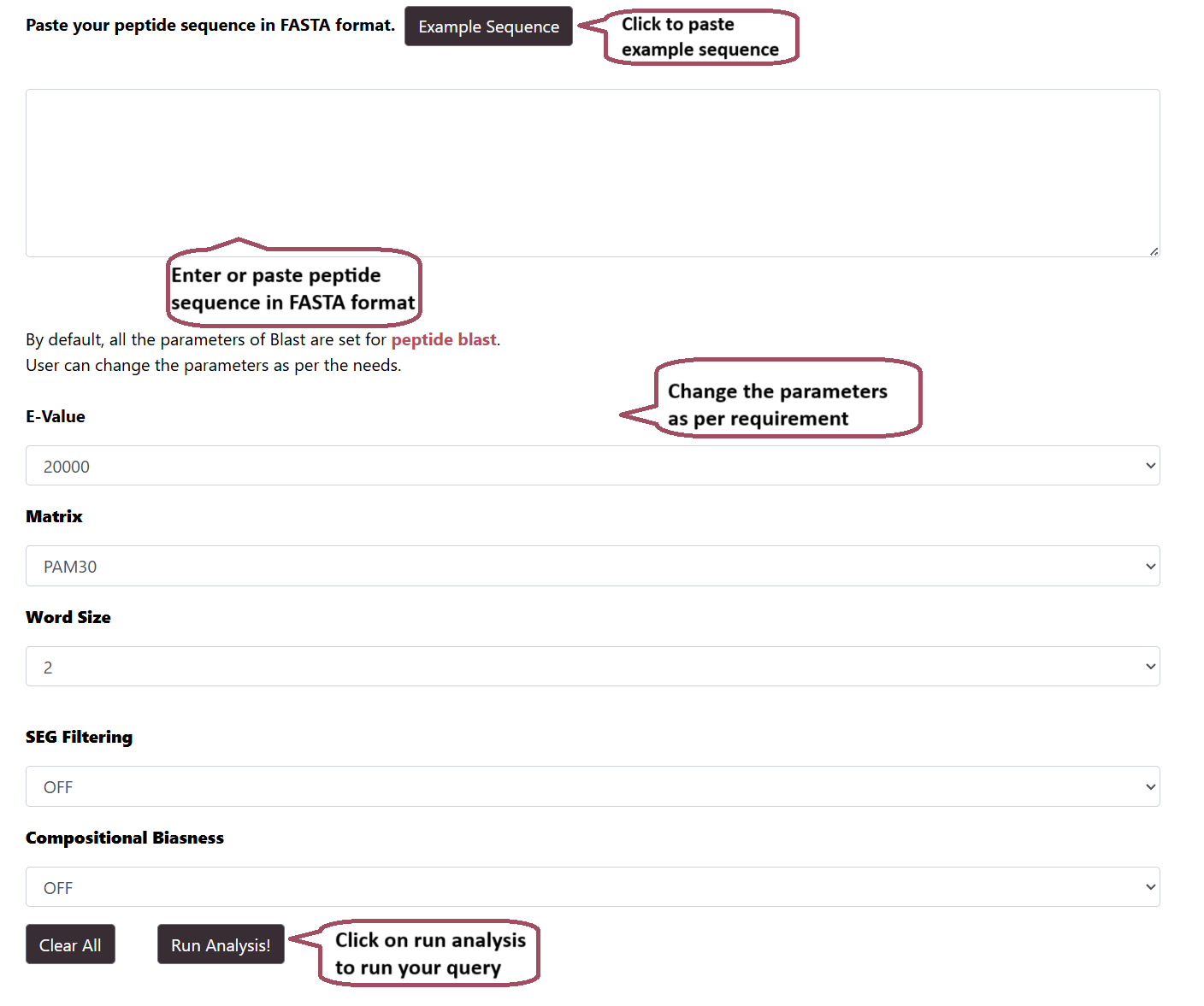
BLAST
Users can run a BLAST query against the Hemolytik 2.0 database. After submission of job it returns the list of peptides similar to the query peptide. The server also provides options to choose different parameters like weight matrix and expectation value.
BLAST Result

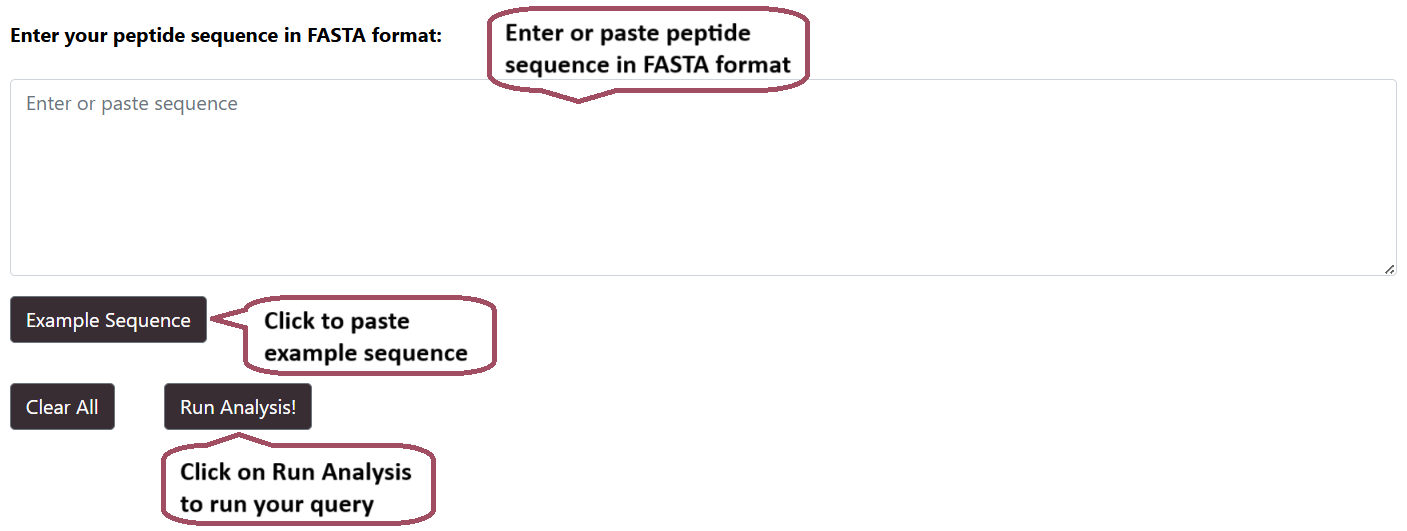
Smith-Waterman Search
Users can run a Smith-Waterman search query against the Hemolytik 2.0 database. After submission of job it returns the list of peptides.
Smith-Waterman Result

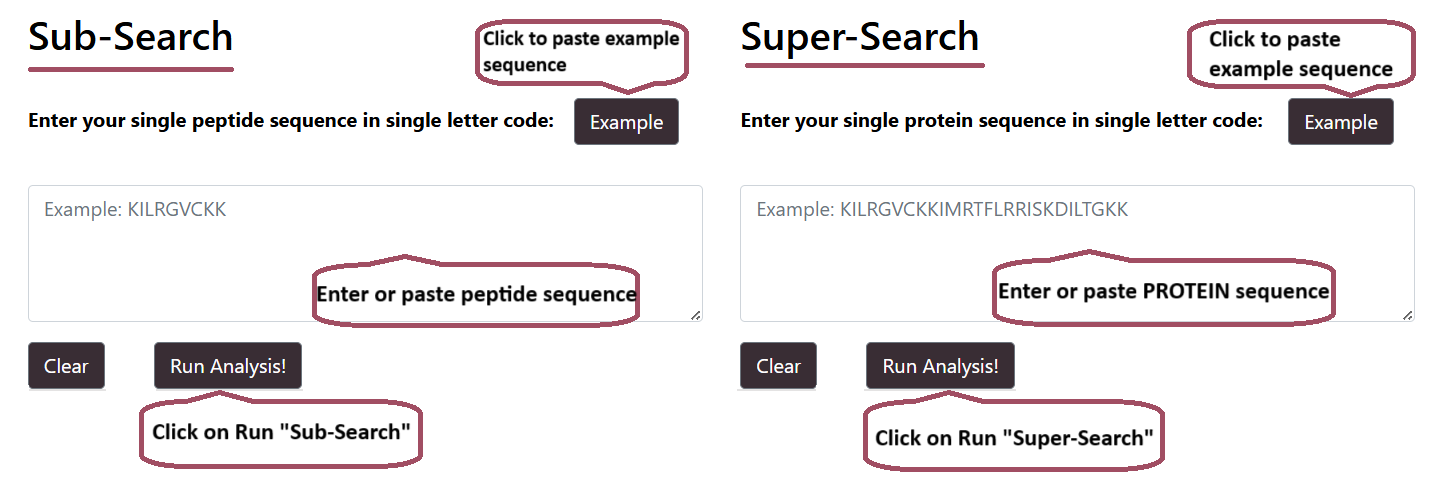
Mapping
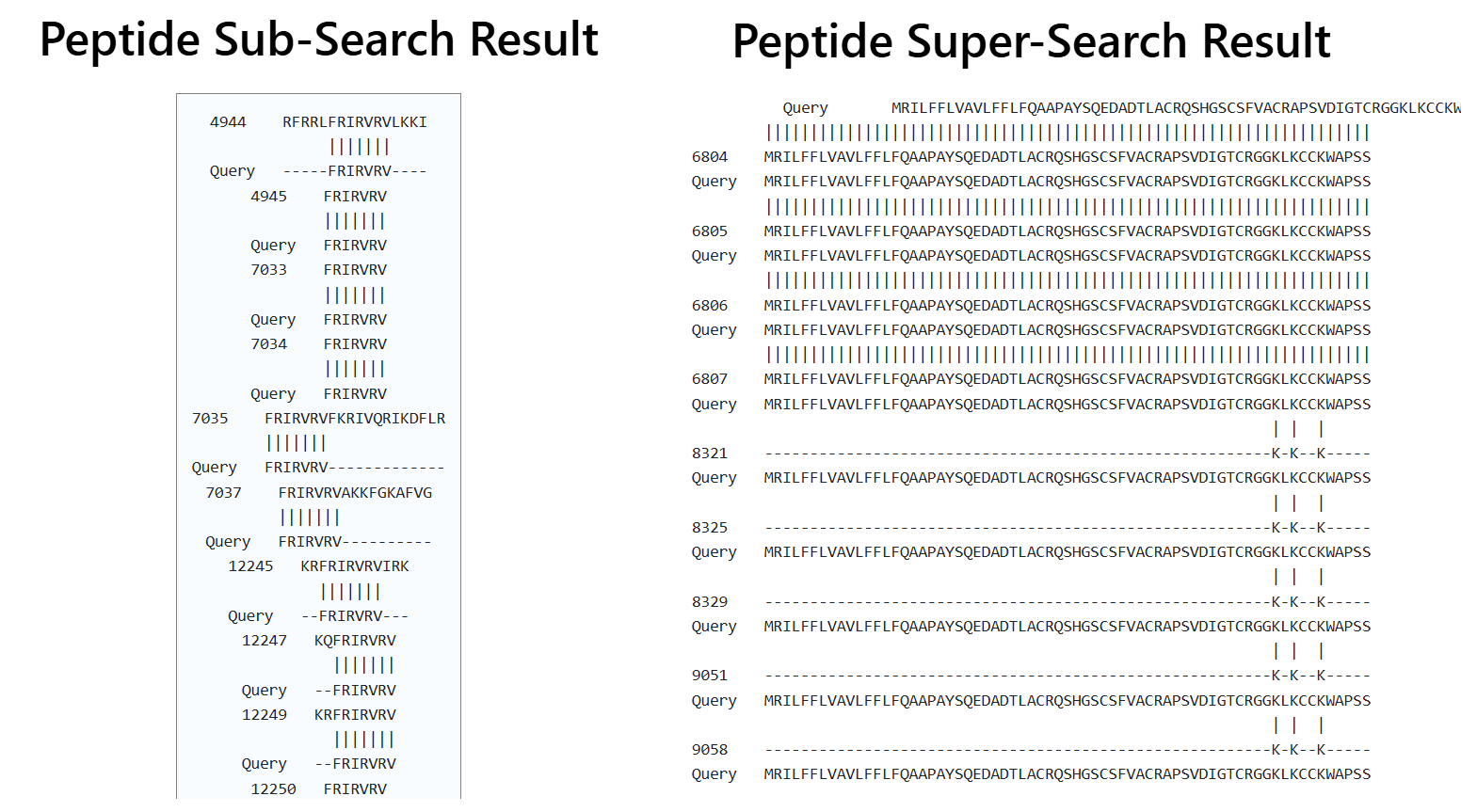
User can select either SuperSearch to search for query PROTEIN sequence against peptides of Hemolytik 2.0 or select SubSearch to search for query PEPTIDE sequence against the peptides of Hemolytik 2.0.
Mapping Result
Structure Alignment
User can align their PDB structure with any of the Hemolytik 2.0 Structures
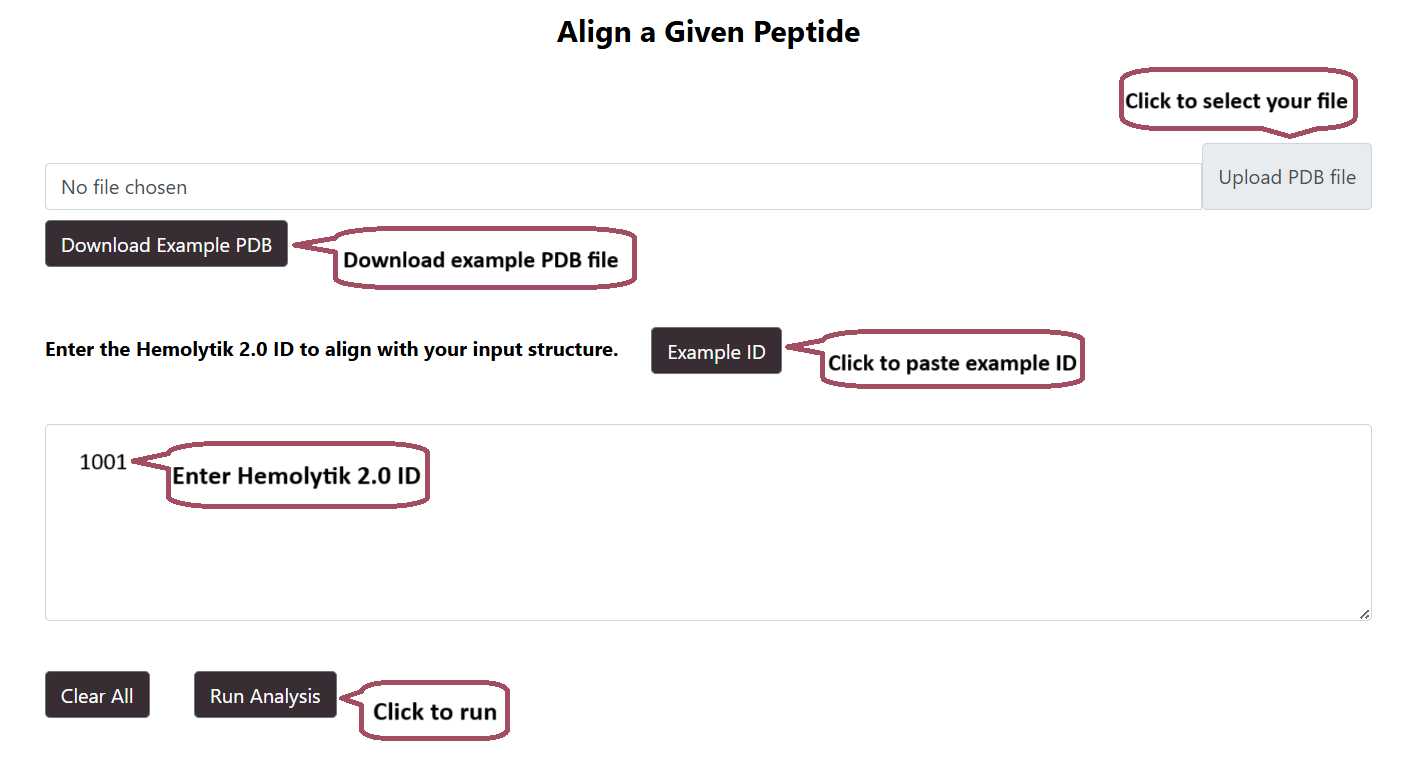
Alignment Result

API
This section describes how this website's data can be accessed with programs. A variety of data
available on Hemolytik 2.0 is accessible using simple URLs (REST) that can be used in
programs.
The Hemolytik 2.0 REST API returns the response in JSON (JavaScript Object
Notation) format. Users can parse the JSON format to suit their requirements.
HTTP Status Headers
Upon sending a request to the server the following HTTP response headers are returned by the Hemolytik 2.0 REST API:
| Code | Description |
|---|---|
| 200 | The request was processed successfully. |
| 400 | Bad Request. Invalid data type. |
| 404 | Not Found. The requested data doesn't exist. |
| 500 | Internal server error. Most likely a temporary problem, but if the problem persists please contact us. |
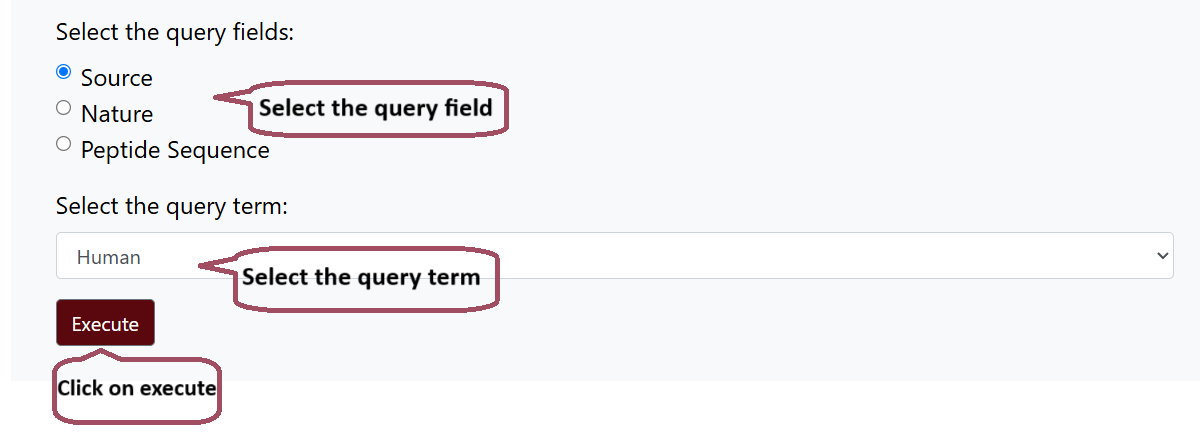
Query Fields
The Hemolytik 2.0 REST API offers access to the data through three distinct query fields: source, nature, and peptide sequence. Users can select from a diverse array of options within each field, enabling targeted retrieval of relevant data. Specifically, the nature query field encompasses 7 distinct nature parameters, comprising data from 20 different Sources and Sequence, which offers two options Natural and Modified. Furthermore, the peptide sequence query field allows users to refine their search based on two parameters: Natural or Modified.
| Query Field (..dataType) | Parameter (..dataValue) | Description |
|---|---|---|
| Source | Human, Rat, Mouse, Fish, Sheep, Rabbit, Horse, Pig, Chicken, Porcine, Mus musculus, etc. | User can access data corresponding to particular source, this will return all the entries for the selected source. |
| Nature | Antimicrobial,Anticancer, Antifungal, Cytotoxic,CPP, etc. | User can access data corresponding to particular nature, this will return all the entries for the selected nature. |
| Peptide Sequence (seq) | Natural, Modified | Users can access data based on their selectedq parameter. Selecting "Natural" will retrieve entries featuring sequences composed solely of natural amino acid residues, and devoid of any chemical modifications. Conversely, selecting "Modified" will retrieve entries characterized by sequences containing non-natural residues or having any chemical modifications. |
Return Fields
Once the request has been processed successfully, the Hemolytik 2.0 REST API returns the data in JSON format. The response data consists of different fields:
| Return Field | Description |
|---|---|
| ID | Unique identifier in the Hemolytik 2.0 database for that entry. |
| PMID | Article PMID corresponding to that entry. |
| Year | Year of publication of that entry. |
| Sequence | Sequence of the peptide. |
| Name | Name of the peptide. |
| C-Ter Modifications | Whether the C-terminal end of the peptide contains an entity or it is free. |
| N-Ter Modifications | Whether the N-terminal end of the peptide contains an entity or it is free. |
| Linear/Cyclic | Conformation of the peptide. |
| Stereo-chemistry | Stereo-chemistry of the peptide. |
| Chemical Modifications | Whether the peptide contains any non-natural residues or any other chemical modifications. |
| Length | Length of the peptide. |
| Nature | Nature of the peptide. |
| Source | The source of peptide used to measure the activity of peptide. |
| Origin | Origin of the peptide corresponding to that source. |
| Experimental Structure | The experimental structure of the peptide. |
| Non-Hemolytic | Whether the peptide is hemolytic or non-hemolytic. |
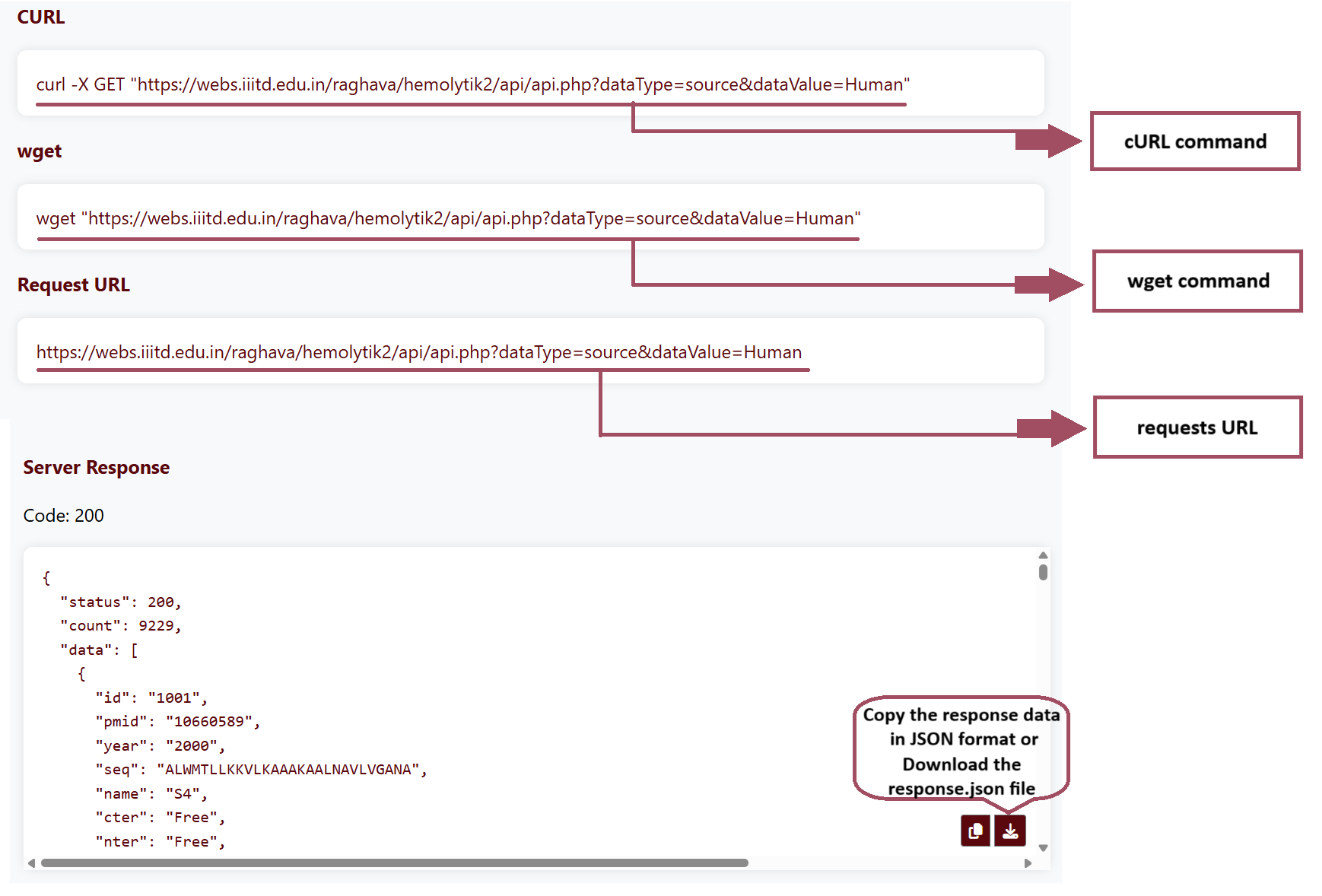
CURL
cURL, short for "Client URL," is a command-line tool and library for transferring data with
URLs. It supports a wide range of protocols, including HTTP, HTTPS, FTP, FTPS, SCP, SFTP,
LDAP, TFTP, and many others. Example Command:
curl -X GET "https://webs.iiitd.edu.in/hemolytik2/api/api.php?dataType=source&dataValue=Human"-X flag is used to specify the HTTP request method. HTTP requests typically use methods such as GET, POST, PUT, DELETE, etc. Hemolytik 2.0 REST API allows only GET request method.
wget
wget is a command-line utility for downloading files from the web. It supports downloading
files via HTTP, HTTPS, and FTP protocols in Linux environments. Example Command:
wget "https://webs.iiitd.edu.in/raghava/hemolytik2/api/api.php?dataType=source&dataValue=Human"
How to use ?
To access the data programmatically using Hemolytik 2.0 REST API, user simply needs to select the query field and its corresponding query term and hit "Execute".
After clicking "Execute" the output will contain the cURL command, wget command, request URL and the server response.
Python Example
Below is a very simple example of how to use the Hemolytik 2.0 REST API using Python. It sends a GET request to the API endpoint, retrieves the JSON response containing data, and parses it into a pandas DataFrame for further analysis. This example demonstrates the basic process of accessing data from the Hemolytik 2.0 REST API and manipulating it within a Python environment.
import requests
import pandas as pd
# Define the URL for the API request
url = 'https://webs.iiitd.edu.in/raghava/hemolytik2/api/api.php?dataType=source&dataValue=Human'
# Send a GET request to the API
response = requests.get(url)
# Check if the request was successful (status code 200)
if response.status_code == 200:
# Print the JSON response from the API
print(response.json())
# Parse the JSON data and store it in a pandas DataFrame
json_data = response.json()
df = pd.DataFrame(json_data['data'])
# Visualize the DataFrame
print(df.head()) # Display the first few rows of the DataFrame
else:
# Print an error message if the request was not successful
print('Error:', response.status_code)