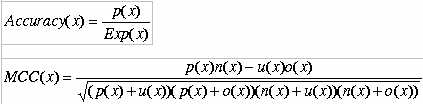Menu |
Algorithm
Evaluation of the predictive performances Support vector machine
The SVM was implemented using freely downloadable software package SVM_light written by Joachims (20). The software enables the user to define a number of parameters as well as to select from a choice of inbuilt kernal functions, including a radial basis function (RBF) and a polynomial kernal. Preliminary tests show that the radial basis function (RBF) kernel gives results better than other kernels. Therefore, in this work we use the RBF kernel for all the experiments. The prediction of functional class is a multi-class classification problem. We developed a series of binary classifiers to handle the multi-classification problem. We constructed N SVMs for N-class classification using 1 vs r (one against rest) strategy. Here, the class number was equal to five forneurotoxin source and function, The ith SVM was trained with all samples in the ith class with positive labels and all other samples with negative labels. In this way, five SVMs were constructed for functional class of neurotoxin source to eubacteria, cnidaria, mollusca, arthropoda and chordata anf function to blocks ion channels, blocks acetylcholinerelease. Protein featuresAmino acid composition.Amino acid composition is the fraction of each amino acid in a protein. The fraction of all 20 natural amino acids was calculated using the following equations: 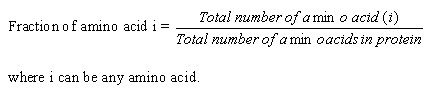
Dipeptide composition. Dipeptide composition was used to encapsulate the global 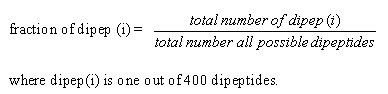
Amino acid composition and length information. The input vector is amino acid composition (20) and the sequence length(1) . The log of length is added as an additional vector to the amino acid composition. PSI-BLASTA module of PSI-BLAST was designed in which query sequences in test dataset were searched against proteins in training dataset using PSI-BLAST. Three iterations of PSI-BLAST were carried out at a cut-off E-value of 0.001. PSI-BLAST was used instead of normal standard BLAST because PSI-BLAST has the capability to detect remote homologies. The module could predict any of the five source (eubacteria, cnidaria, mollusca, arthropoda and chordata) and function (blocks ion channels, blocks acetylcholine receptors, inhibits acetylcholine release by metalloproteolytic activity and phospholipase A2 and facilitates acetylcholine release) and sub-classification of blockers of ion channels(Sodium, Potassium, Calcium and Chloride) depending upon the similarity of the query protein to the protein in the dataset. |