Modification and Annotation in Proteins (MAP)
The Modification and Annotation in Proteins (MAP) format is a new way to represent protein sequences, going beyond the traditional FASTA format. While FASTA only supports the 20 standard amino acids, MAP allows scientists to add extra information like:
- Modified or Unusual Amino Acids
- Post-translational Modifications
- Binding Sites (like where a protein interacts with DNA or metal ions)
- Mutations (substitutions, insertions, deletions)
- Protein Metadata (such as function, organism, location)
MAP keeps the sequence human-readable and software-friendly by using {curly brace tags} to embed this information directly into the protein sequence or header line.
This makes MAP perfect for researchers who want to include both sequence and detailed biological context — all in one place. Learn More About MAP Format.
Shendre, Akshay, et al. "MAP Format for Representing Chemical Modifications, Annotations, and Mutations in Protein Sequences: An Extension of the FASTA Format." ArXiv, 2025, https://arxiv.org/abs/2505.03403
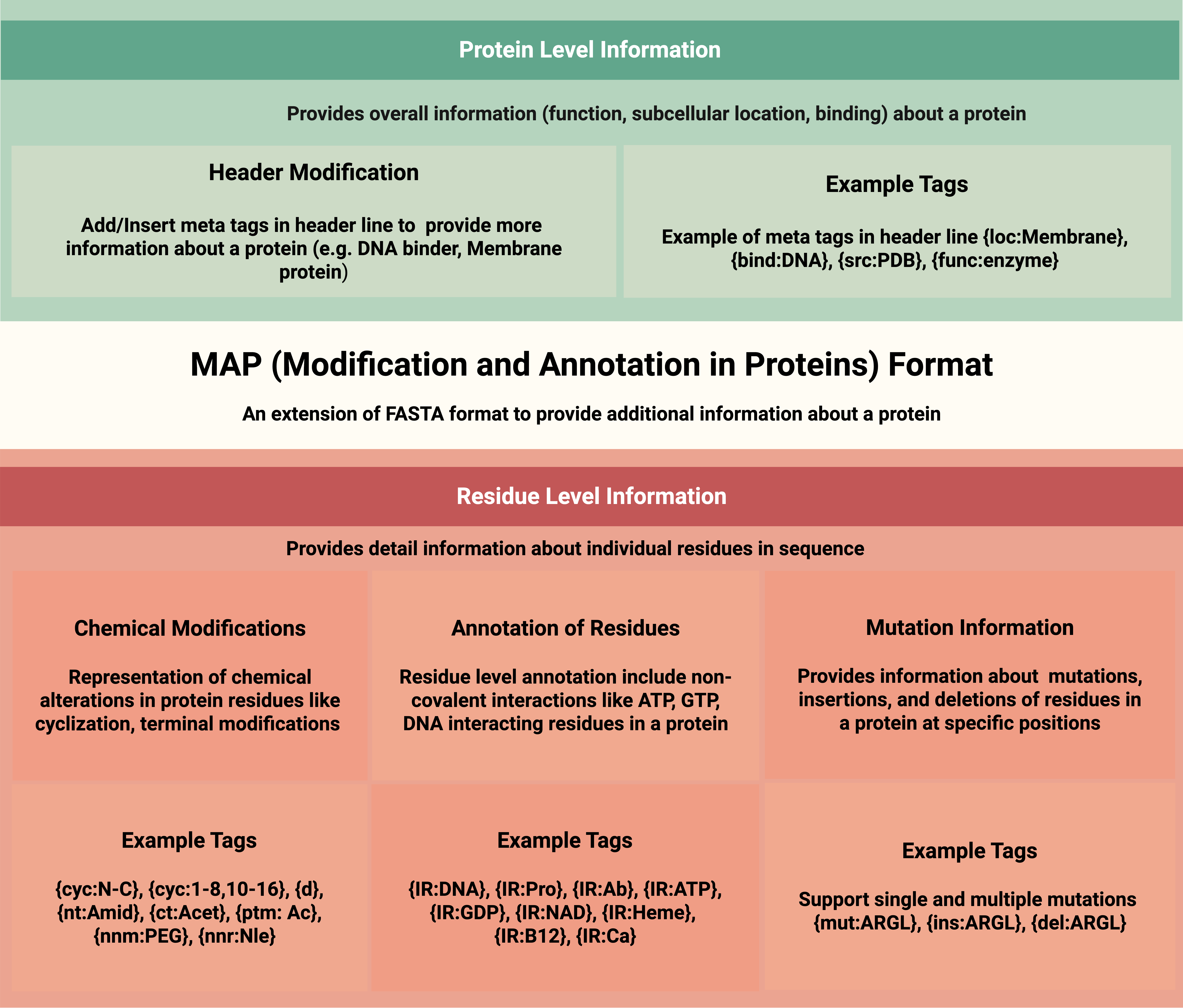
Figure 1: Overview of the MAP Format
Explore the computational tools and algorithms designed to unlock the power of the MAP format.
This outlines our algorithmic pipeline for effectively handling and analyzing protein data in the enriched MAP format. From seamless format conversions to advanced machine learning for property prediction, discover the tools that empower your research.
Feature Extraction (mFeatures)
Extract meaningful features from MAP sequences and annotations.
Discover »