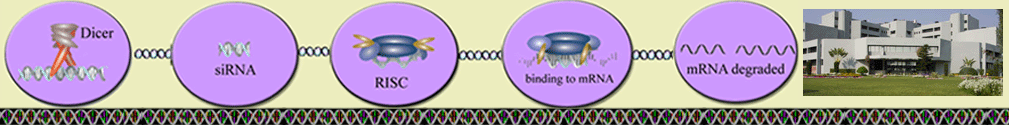
Algorithm
Dataset:
We have chosen the largest dataset of 2431 siRNAs (21mer) derived from a homogeneous experimental condition as reported by Huesken
et al termed hereafter as Main21 dataset. In this study, out of 2431 siRNAs, 2182 were used for developing our models and rest 249 used as independent dataset for testing our models. Since this dataset comprises of 21mer siRNAs having two deoxyribonucleotide base-pair 3' overhang, a direct comparison between 21mer and 19mer siRNAs could not be made and hence for comparison sake a new dataset Main19 of 19mer siRNAs was created after removing the two base pair overhang from 3' end, from original Huesken's 21mer siRNAs. We also used another commonly used datset comprising of 581 siRNAs (19mer) sequences derived from heterogenous experimental conditions reported by Seatrom
et al, hereafter denoted as Alternate19 dataset.
Independent Datasets:
In the present study several datasets representing experimentally validated siRNAs targeting different genes were used to evaluate the performance of our SVM models. Heusken's reported 249 test siRNAs were used as an independent dataset to evaluate the performance of SVM models developed on Main21 and Main19 datasets. Besides that dataset of 526 siRNA reported by Holen and dataset of 653 siRNAs complied by Shabalina and its subset of 156 siRNAs not included in Saetrom dataset were also taken for efficacy evaluation.
SVM:
Support Vector Machine (SVM) supports both classification and regression tasks and can handle multiple, continuous and categorical variables. The selection of kernel is very important in SVM, which is analogous to choosing architecture in ANN. SVM contains all the main features that characterize maximum margin algorithm: a non-linear function is learned by linear learning machine, mapping into high-dimensional kernel induced feature space. The capacity of the system is controlled by parameters that do not depend on the dimensionality of feature space. In a regression SVM, we estimate the functional dependence of the dependent variable Y on an independent variable X. In this study nucleotide frequencies are used as independent variable and efficacy of siRNA as dependent variable. We implemented SVM using SVM_light package, which allows choosing number of parameters and kernels e.g. linear, polynomial, radial basis function (RBF) kernel.
Parameters used from siRNA sequence: In total thirteen different parameters were employed including five nucleotide frequencies, one binary pattern and seven hybrid approaches based on above parameters
Nucleotide Frequencies:
The objective of calculating nucleotide frequency of siRNA sequences is to know occurrence of mononucleotide, dinucleotide, trinucleotide, tetranucleotide and pentanucleotide subsequences in a given siRNA sequence. his can transform any length of nucleotide sequence to a fixed length feature vectors. It is important while using machine-learning technique because it requires fixed length input patterns. The information of each siRNA can be encapsulated to a vector of 4, 16, 64, 256 and 1024 multi-dimensions using frequencies of its mononucleotide, dinucleotide, trinucleotide, tetranucleotide and penta-nucleotide subsequences respectively. Nucleotide strings above penta-nucleotide were not considered since representation of these strings decreased considerably in 21mer siRNA sequences.
Binary Pattern:
We employed binary pattern to extract siRNA features based on the occupancy of nucleotides at each position of siRNA sequences. Four binary patterns used for each nucleotide are follows a=1 0 0 0; c=0 1 0 0; g=0 0 1 0 and t/u=0 0 0 1 and this resulted in accumulation of 84 and 76 patterns for each of 21-mer and 19-mer siRNA.
Hybrids Approaches:
In hybrid approach, different combinations of mono to penta nucleotide frequencies and binary pattern were used in order to increase the performance of the prediction methods. We have developed total seven hybrids methods (hybrid-1 to hybrid-7) as shown in Table 1. To develop best model (Hybrid-7), a total of 1448 SVM input patterns were used for 21mer siRNA sequence.
Evaluation:
We computed the correlation between predicted and actual efficacy of siRNA, in order to assess the performance of our models. In this study, we used 10-fold cross-validation where dataset was randomly divided into 10 equal sets; 9 sets were used for developing model and remaining set for testing, this process is repeated 10 times in such a way that each set is used once for testing. Ten fold cross validation technique was executed on Main21, Main19 and Alternate19 datasets.
Pearson correlation coefficient (or correlation as used in this study) between the actual and the predicted siRNA efficacies was calculated to assess the performance of our real efficacy prediction modules. The Pearson correlation coefficient (PCC) between the actual and the predicted efficacies has been calculated using following equation.
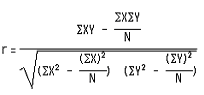
Where R is the Pearson's coefficient of correlation, X denotes the actual efficacy values and Y is the predicted efficacies. 'N' here is the total number of examples.