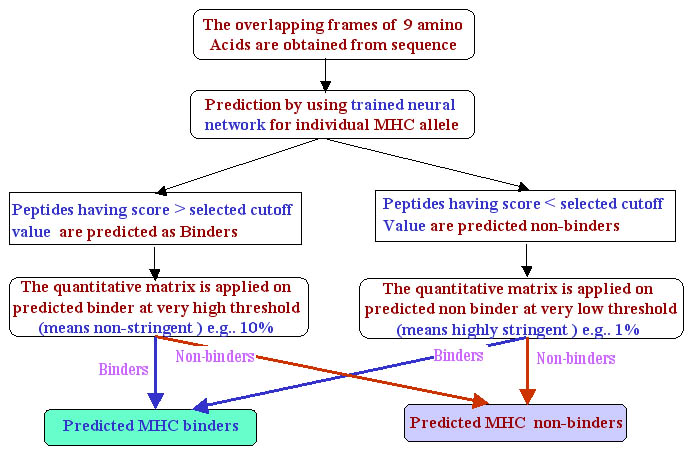
Generation of Quantitative matrices:-
The quantitaive matrices consist of a table having the sequence weight frequencies of each of the 21 amino acids (including "X") at each position in the dataset of MHC binders divided by the corresponding expected frequency of that amino acid in the non-binders dataset. The MHC binders datasets for each MHC allele are generated by obtaining MHC binders of 9 amino acids from MHCBN database. The equal number of the non-binders is also obtained from the same database (if available) otherwise the 9-mer peptides are randomly choosen from the SWISS-PROT database. The quantitative matrices are addition matrices where the score of a peptide is calculated by summing up the scores of each residue at specific position along peptide sequence. For example, the score of peptide "ILKEPVHGV" is calculated as follows.
Score= I(1)+L(2)+K(3)+E(4)+P(5)+V(6)+H(7)+G(8)+V(9) (1)
The peptides with score more than the cutoff score at a particular threshold are predicted as MHC binders.A few matrices are also obtained from literature(BIMAS and ProPred1).These matrices are mostly multiplication matrices. The score of the peptide is calculated as follows:e.g "ILKEPVHGV"
Peptide score=I(1) * L(2) * K(3) * E(4) * P(5) * V(6) * H(7) * G(8) *V (9) (2)
Threshold score: The determination of threshold or cutoff score is an integral part of matrices based predictions. It is prerequisite to calculate the cutoff score for each of the matrices. The calculation of the threshold score requires sufficient amount of data of MHC binders and non-binders. The threshold score for the matrix of each MHC allele is determined as follows:
Firstly, all overlapping peptides of 9 amino acids are generated from all the proteins present in SWISS-PROT database.
Secondly, the score for these natural SWISS-PROT peptides are obtained by using new quantitative matrices of different MHC alleles. These peptides are sorted in the desending order depending on the score achieved by each peptide. The top 1% of the peptides are extracted and the minimum score out of these peptides is considered as threshold score at 1%. Similarly, the peptide scores at other thresholds such as 2%,3% ,etc. is also calculated.
The peptide getting score more than cutoff score at particular threshold are known as binders. On the otherhand the peptide getting score less than threshold score are known as non-binders.