Top
NAGbinder Help Page
This is a help page of NAGbinder, developed for predicting NAG binding sites in a protein. This page provides diffrent type of information on NAGbinder. In order to provide information in structured forms, we have divided information in following topics.
- Importance of NAGbinding: N-Acetylglucosamine (NAG) is one of the eight crucial sugars required to maintain the optimal health and functioning of the human body.
- Datasets: Datasets were generated from NAG binding protein structure in PDB (April 2019, release).
- Evaluation of models: Standard protocols were used for evaluating models develop in this study.
- Algorthm: Standard algorithm are used for devloping NAG interacting residues.
- Help on Web Pages: Help pages with screen shots
Goto Top
Goto Top
Goto Top
Goto Top
Goto Top
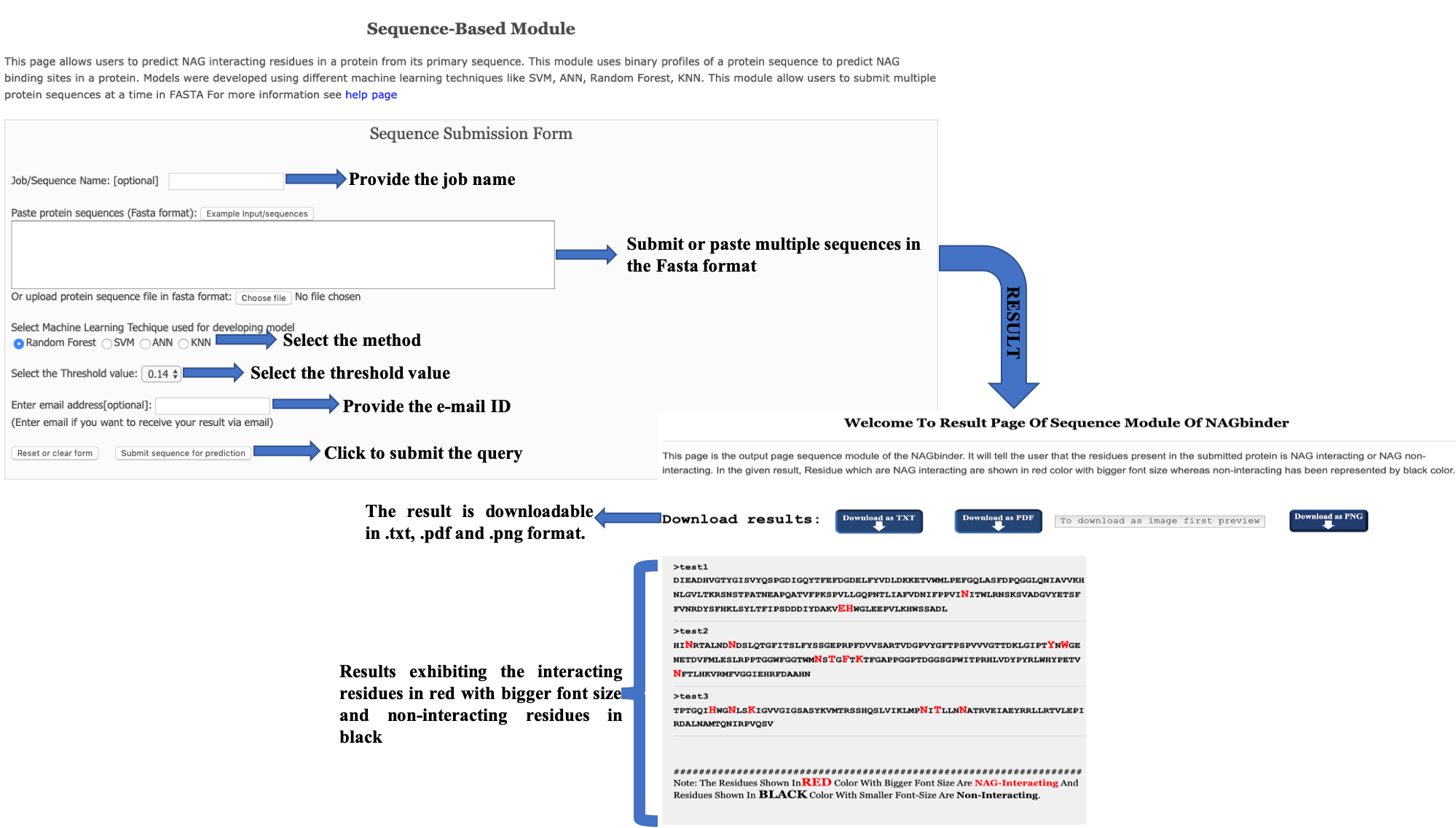
Help PagesNAGbinder server discriminate the NAG interacting residues and non-interacting residues from a given sequence. The NAGbinder server uses the SVC based method by using the Position Specific Scoring Matrix (PSSM) generated from the query sequence(s). The overall accuracy of this server is ~ 96.00%. NAGbinder is a web-server specially trained for the NAG interacting residues. The prediction is based on the basis of PSSM pattern of 9 window motif of amino acid sequence by using support vector classifier (SVC). Probability score is calculated in between 0-9 and higher the probability value, higher is the chances of the residue to be NAG interacting. |
|
This page provides help on different modules of server. Following are major modules in this server:
|