![]()
• ESLpred Home
• Submit Protein
• Help
• Algorithm
• Developers
• Contact
Other Servers
General Information & Stepwise Help
General Information:-
In this era of genomics, the raw sequence data is being produced at exponential rate. This had created the need for automated genome annotation methods so that biological information can be obtained from sequence information.The determination of cellular localization of protein can provide important clue to elucidate the function of protein. The development of improved and efficient sub-cellular localization prediction methods can assist in the analysis of piled genomic data.In the past, many efforts have been made to develop computational methods for predicting the localization of proteins in the cells using different approaches.These methods are mostly based
- Homology to some protein of known function.
- Sequence motifs.
- N-terminal sorting signals.
- Composition of amino acids.
- PSORT I and PSORT-B for prokaryotic organisms.
- iPSORT and TargetP for eukaryotic organisms.
- NNPSL and SubLoc for both prokaryotic and eukaryotic organisms.
Stepwise Help:-
Name of Protein-: This is an optional field.The name of protein may have letters and number with the "-" or "_". All other character are non-permissible.The field is assigned a default name "Protein". The sequence name is just used for only your information. It may be a problem with ?, ?, ? for example or an empty space within the name of the sequence, which is not allowed for reasons of security.
Protein sequence-:This server allows the submission of sequence in any of the standard formats. The user can paste plain sequence in the provided inbox.The server also has the facility for uploading the local sequence files. Amino acid sequences must be entered in the one-letter code.All the non standard characters will be ignored from the sequence.
Sequence Format:-The server can accept both the formatted or unformatted raw antigenic sequences.The server uses ReadSeq routine to parse the input.The user should choose wether the sequence uploaded or pasted is plain or formatted before running prediction.The results of the prediction will be wrong if the format choosen is wrong.
Prediction Approach:-The method allow the prediction on the basis of five different appraoches. These approaches along with the reliability to predict the subcellulat localization of proteins.The prediction is based on
- Composition of amino acids:- A SVM was developed on the basis of composition amino acids of protein. The SVM was provided with a 20 dimensional vector. The amino acid composition is fraction of each amino acid in a protein. The ovaerall accuracy of composition based method for four subcellular locations (Nuclear,Cytoplasm,Mitochondria and Extracellular) is 78.1%. The performence of the method is evalvuated using five fold cross-validation.
- Physico-chemical properties of amino acids:- A SVM was developed on the basis of avearge physiochemical properties of protein sequence. The properties of protein was determined by considering 33 physico-chemical properties. The list of the properties along with bibliographic information is provided in table 1 ( supplymentary material). The ovaerall accuracy of properties based method is 77.8%.
- Composition of Dipeptides:- A SVM was developed on the basis of composition of dipeptides of protein sequence. This will give a fixed pattern length of 400. This representation encompasses the information about amino acid composition along local order of amino acid. The ovaerall accuracy of properties based method is 84.6%.
- EuPSI-BLAST:- The subcellular localization tends to be evolutionary conserved. so homology to a protein of known localization appears to be a good indication of protein actual localization. Therefore, we have constructed a module EuPSI-BLAST, in which the PSI-BLAST search of query sequence is carried out against a database of 2427 eukaryotic proteins. The PSI-BLAST was used for three iteration at a cutoff Evalue of 0.001.The module predicts nuclear, cytoplasmic, mitochondrial and extracellular protein with 84.5%, 77.6%, 54.8% and 86.7% accuracy respectively.
- Hybrid Method:- To improve the accuracy of prediction we have adopted many different strategies for encapsulating the global information about a protein. The hybrid SVM module for each localization was developed on the basis amino acid composition, dipeptide composition, physico-chemical properties and BLAST output. The hybrid module was provided with a input vector of 458 dimensions including 20 for amino acid composition, 400 for dipeptide composition, 33 for physiochemical properties and five for blast output. The ovaerall accuracy of properties based method is 88.0%.
- Nakai, K. and Kanehisa, M. (1991) Expert system for predicting protein localization sites in gram-negative bacteria. Proteins, 11, 95-110.
- Nakai, K. and Horton, P. (1999) PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci. 24, 34-6.
- Bannai, H., Tamada, Y., Maruyama, O., Nakai, K. and Miyano, S. (2002) Extensive feature detection of N-terminal protein sorting signals. Bioinformatics, 18, 298-305.
- Emanuelsson, O., Nielsen, H., Brunak, S. and von Heijne, G. (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol., 300, 1005-16.
- Reinhardt, A. and Hubbard, T. (1998) Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res., 26, 2230-6.
- Hua, S. and Sun, Z. (2001) Support vector machine approach for protein subcellular localization prediction. Bioinformatics, 17, 721-8.
- Bairoch, A. and Apweiler, R. (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res., 28, 45-8.
- Gardy, J.L., Spencer, C., Wang, K., Ester, M., Tusnady, G.E., Simon, I., Hua, S., deFays, K., Lambert, C., Nakai, K. and Brinkman, F.S. (2003) PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res., 31, 3613-7.
- Joachims, T. (1999) Making large-Scale SVM Learning Practical. In: B Scholkopf and C Burges and A Smola, (eds) Advances in Kernel methods –support vector learning. MIIT Press, Cambridge massachusetts,London England .
- Bhasin, M. and Raghava, G.P.S. (2003) A SVM based method for prediction of HLA-DRB1*0401 binders in antigen sequence. Bioinformatics (In Press).
- Tusnady, G.E. and Simon, I. (2001), The HMMTOP transmembrane topology prediction server. Bioinformatics. 17, 849-50.
- Fujiwara, Y. and Asogawa, M. (2001) Prediction of subcellular localizations using amino acid composition and order. Genome Inform Ser Workshop Genome Inform. , 12, 103-12.
- Bhasin, M. and Raghava, G.P.S. (2003) Analysis and prediction of quantitative affinity of TAP binding peptides using cascade SVM. Protein Science (In Press).
- Zavaljevski, N., Stevens, F.J. and Reifman, J. (2002) Support vector machines with selective kernel scaling for protein classification and identification of key amino acid positions. Bioinformatics, 18, 689-96.
- Brown, M.P., Grundy, W.N., Lin, D., Cristianini, N., Sugnet, C.W., Furey, T.S., Ares, M. Jr. and Haussler, D. (2000) Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci U S A., 97, 262-7.
- Ward, J.J., McGuffin, L.J., Buxton, B.F. and Jones, D.T. (2004) Secondary structure prediction with support vector machines. Bioinformatics, 2003, 19, 1650-5.
An Sample of Query Submission form is shown below
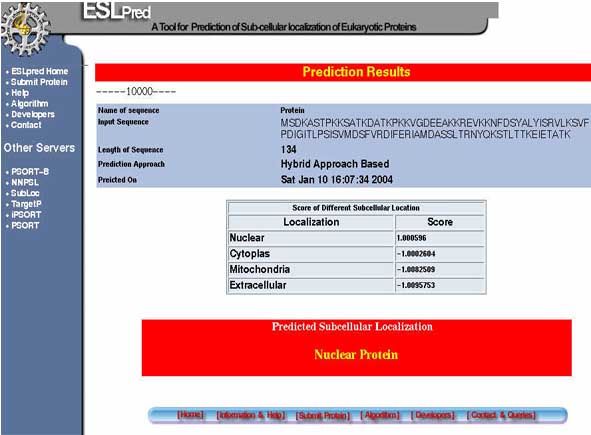
Prediction Results:-After the analysis the result having the sequence of protein, length of protein, score of protein for each subcellular localization is illustrated. The results also depict the final predicted sub-cellular localization of protein.A sample of results page is shown below.