
![]()
• ESLpred Home
• Submit Protein
• Help
• Algorithm
• Developers
• Contact
Other Servers
Detailed Algorithm & Dataset Information
Dataset Information:- The dataset used for training in this paper was the same as of NNPSLand SubLoc. The dataset was generated from the 33.0 version of SWISS-PROT by Reinhardt and Hubbard. The dataset have the protein which are complete and whose subcellular localization is experimentally determined. The dataset was redundancy reduced such that none protein have >90% sequence identity to any other protein in dataset. We have only obtained the dataset of 2427 eukaryotic proteins. This dataset consists of . 1097 nuclear, 684 cytoplasmic, 321 mitochondrial and 325 extracellular proteins.
Support Vector Machine
The support vector machines (SVM) are universal approximator based on statistical and optimising theory. The SVM is particularly attractive to biological analysis due to its ability to handle noise, large dataset and large input spaces. The SVM has been shown to perform better in protein secondary structure, MHC and TAP binder prediction and analysis of microarray data. The basic idea of SVM can be described as follows; first the inputs are formulated as feature vectors. Secondly these feature vectors are mapped into a feature space by using the kernel function. Thirdly, a division is computed in the feature space to optimally separate to classes of training vectors. The SVM always seeks global hyperplane to separate the both classes of examples in training set and avoid overfitting. The hyperplane found by SVM is one that maximise the separating margins between both binary classes This property of SVM is made is more superiors in comparison to other classifiers based on artificial intelligence.The basic idea of SVM is depicted below
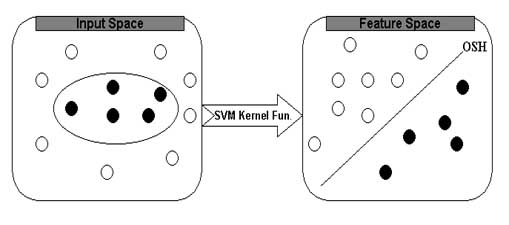
In this study, we have used SVM_light to predict the subcellular localization of proteins. The software is freely downloadable from http://www.cs.cornell.edu/People/tj/svm_light/. The software enable the users to define a number of parameters and allow to select a choice of inbuilt kernel function including linear, RBF, Polynomial (given degree) or user defined kernel.
For classifying the proteins to particular localization (out of 4), we have constructed four SVM known as cytoplasmic, Nuclear, extracellular ands Mitochondrial SVM modules. Each SVM module is Binary SVM that produces a single score for each of protein. A particular SVM (for example Cytoplasmic) will be trained with all samples of cytoplasmic class as positive label and rest samples with negative lable. The SVM modules for nuclear, extracellular and mitochondrial proteins are also trained similarly.
Evaluation of Modules:- The performance of all modules developed in this method is evaluated using five-fold cross-validation. In five- fold cross validation dataset was divided into five equal size sets. The training and testing of every module (BLAST and various SVM’s) was carried five times, each time using one distinct set for testing and remaining four sets for training. The performance of each module is assessed by calculating the accuracy and Matthew's correlation coefficient (MCC).The formula's of acuuracy and MCC calculation are shown below.
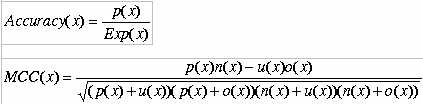
Where x can be any subcellular location (nuclear, cytoplasm, extracellular and mitochondria) exp(x) is the number of sequences observed in location x, p(x) is the number of correctly predicted sequences of location x, n(x) is the number of correctly predicted sequences not of location x, u(x) is the number of under-predicted sequences and o(x) is the number of over-predicted sequences.
Prediction Approaches:- In this study authors have developed modules on the basis of support vector machines (SVM), similiarity search (BLAST)and combination of SVM and BLAST.The SVM based modules were trained with various inputs to encapsulate the global information of protein sequence for more accurate prediction.The various SVM and BLAST modules developed in this study are described below.
- Composition based SVM:-The SVM was provided with 20 dimensional vector on the basis of composition amino acids of protein. The amino acid composition is fraction of each amino acid in a protein. The four SVM's were trained for four localization of eukaryotic proteins.In composition based modules best results are achieved using RBF kernel. The value of Gamma factor and regulatory parameter "C" was optimised to "16" and "1000" respectively.The results obtained after five fold cross-validation are shown below in tabular form.
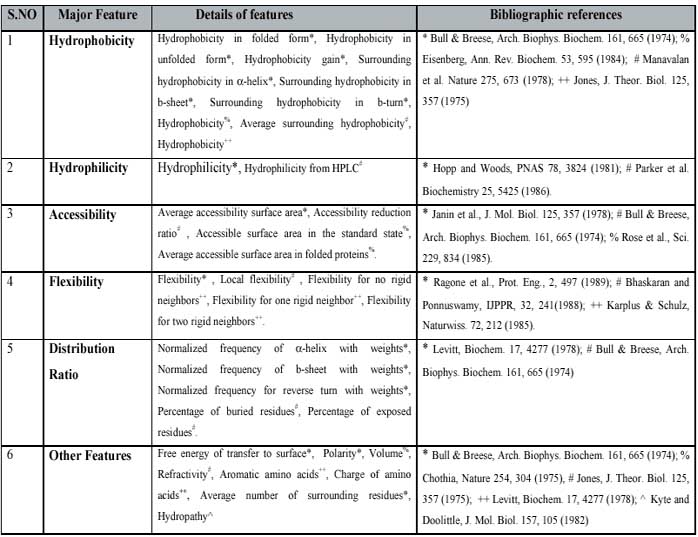
- Physico-chemical properties based SVM:-A SVM was developed on the basis of avearge physiochemical properties of protein sequence. The properties of protein was determined by considering 33 physico-chemical properties. The list of all physico-chemical properties is shown in table S1.The best results are achieved using RBF kernel. The value of Gamma factor and regulatory parameter "C" was optimised to "15" and "1000" respectively.The results obtained after five fold cross-validation are shown below in tabular form.
- Dipeptide composition based SVM:-A SVM was developed on the basis of dipeptide composition of proteins.The global information of protein can be encapsulated to a vector of 400 dimensions using dipeptide composition. The best results are achieved using RBF kernel. The value of Gamma factor and regulatory parameter "C" was optimised to "200" and "1000" respectively.The results obtained after five fold cross-validation are shown below in tabular form.
- EuPSI-BLAST:-we have constructed a module EuPSI-BLAST, in which the PSI-BLAST search of query sequence is carried out against a database of 2427 eukaryotic proteins. The PSI-BLAST was used for three iteration at a cutoff Evalue of 0.001. The PSI-BLAST was used instead of normal standard BLAST because PSI-BLAST is able to find out the proteins having remote homology. The module returns the subcellular localization of protein, SWISS-PROT number and sequence of protein having similarity to query sequence.The performence of the module was evalvuated using five fold cross-validation. During cross-validation no significant hit is obtained for 362 protein. Using EuPSI-BLAST 84.5%, 77.6%, 54.8%, 86.7% accurcies for nuclear, cytoplasmic, mitochondrial and extracellular proteins respectively.
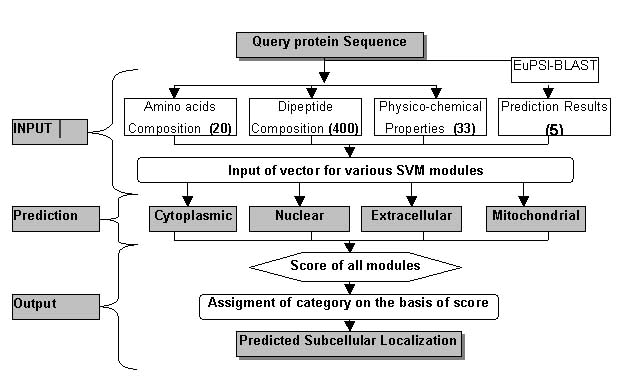
- Hybrid Approach based Prediction:-To improve the prediction accuracy we adopted mmany statergies. In the last, we developed a hybrid approach on the basis amino acid composition, dipeptide composition, physico-chemical properties and BLAST output. The hybrid module was provided with a input vector of 458 dimensions including 20 for amino acid composition, 400 for dipeptide composition, 33 for physiochemical properties and five for blast output.The overall architecture of the hybrid module is shown below.
| Cellular Localtion | Accuracy(%) | MCC |
|---|---|---|
| Nuclear | 86.1 | 0.73 |
| Cytoplasm | 76.9 | 0.64 |
| Mitochondria | 55.5 | 0.54 |
| Extracellular | 76.0 | 0.76 |
| Cellular Localtion | Accuracy(%) | MCC |
|---|---|---|
| Nuclear | 85.6 | 0.73 |
| Cytoplasm | 74.6 | 0.64 |
| Mitochondria | 59.2 | 0.55 |
| Extracellular | 76.6 | 0.74 |
Table S1: list of physico-chemical properties.
| Cellular Localtion | Accuracy(%) | MCC |
|---|---|---|
| Nuclear | 92.7 | 0.79 |
| Cytoplasm | 80.2 | 0.71 |
| Mitochondria | 58.8 | 0.62 |
| Extracellular | 79.0 | 0.83 |
In the hybrid approach best results are obtained with RBF kernel. we have also used the polynomail kernel and linear kernel but the results are poorer in comprasion to RBF kernel.The value of the gamma and regulatory parameter C optimised to "50" and "1000" respectively.The prediction results are shown below in tabular form.
| Cellular Localtion | Accuracy(%) | MCC |
|---|---|---|
| Nuclear | 95.3 | 0.87 |
| Cytoplasm | 85.2 | 0.79 |
| Mitochondria | 68.2 | 0.69 |
| Extracellular | 88.9 | 0.91 |
The systematic increase in prediction accurcy and MCC from only amino acid composition to hybrid approach based module is depicted below.
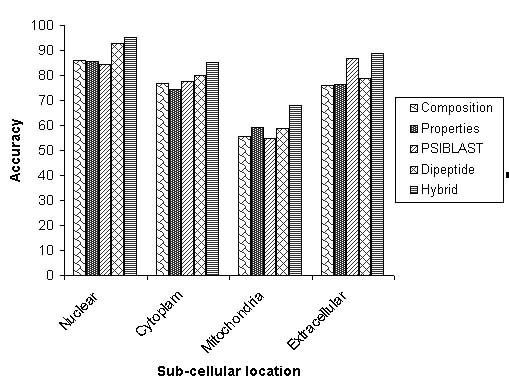
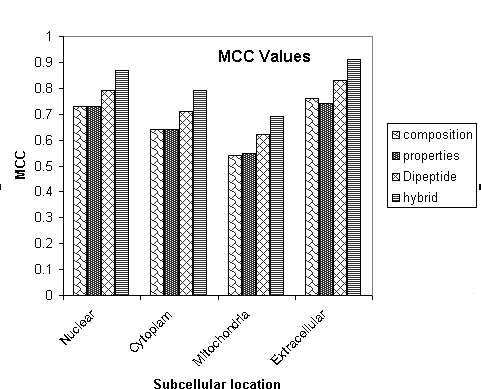
The calculation of reliability index is important to known the reliability of prediction.In this study we have followed the simple statragey of Hua,S. and Sun, Z. (2001) for assigning the reliability index (RI). The RI is assigned on the basis of difference between the highest and second highest output score of SVM's for various subcellular localizations.A curve is plotted in between the prediction accuracy and reliablity index equal to particular value as shown below.
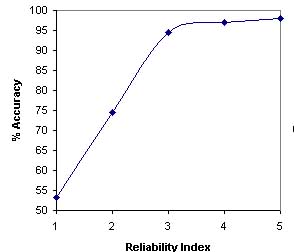
The curve depict that expected accuracy of sequences with RI=3 is 94.4% which is better in comparsion to exiting methods. Another claculation depict the nearly 74% of sequence have RI graeter than 2 and expected accuracy of these sequence is 96.4%.