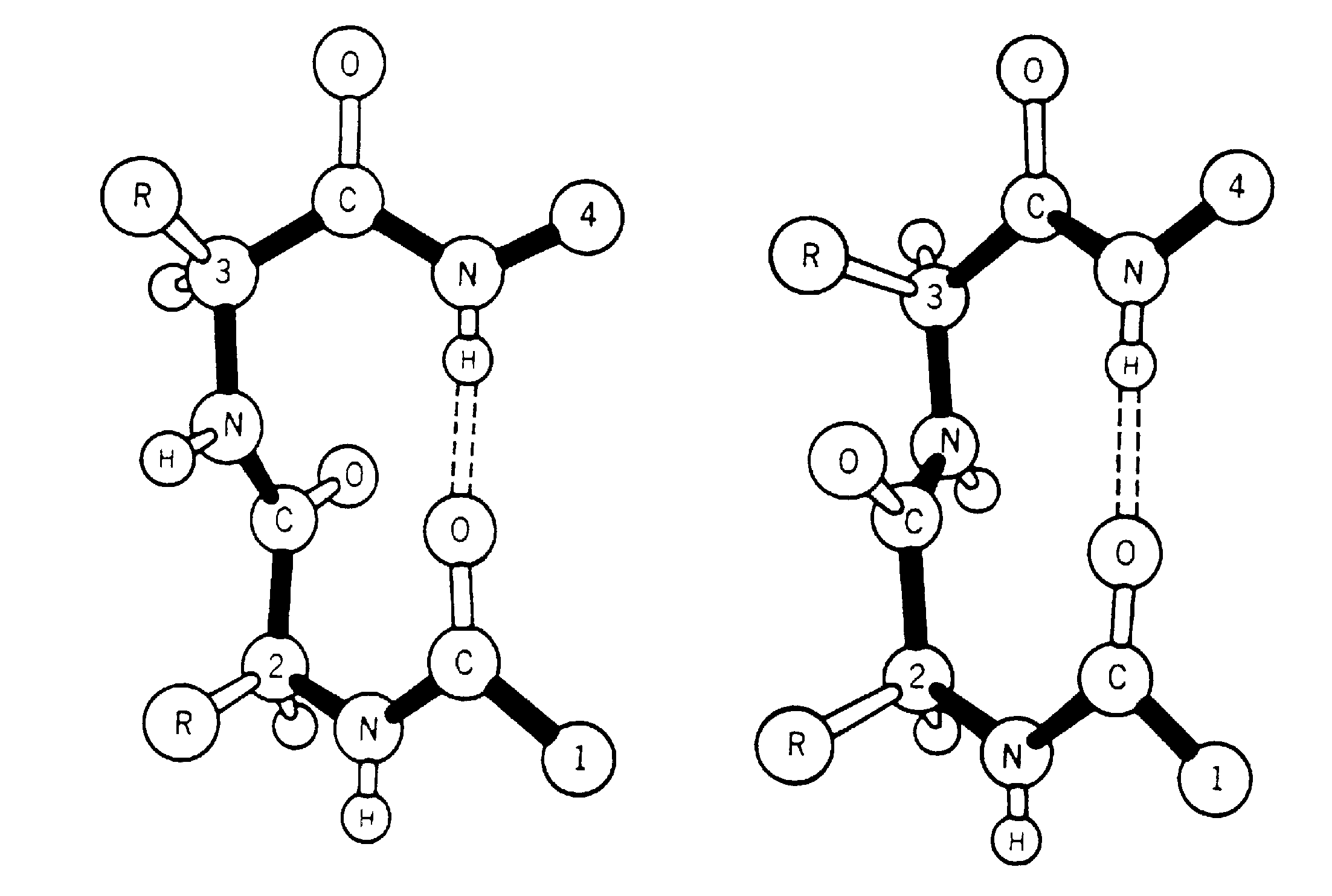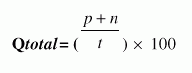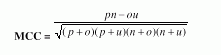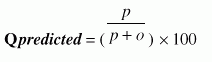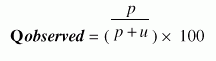



| BetaTPred2: Prediction of ß-turns in proteins using neural networks and multiple alignment |
|
|
|
|
|
|
|
Neural Networks Definition Also referred to as connectionist architectures, parallel distributed processing, and neuromorphic systems, an artificial neural network (ANN) is an information-processing paradigm inspired by the way the densely interconnected, parallel structure of the mammalian brain processes information. Artificial neural networks are collections of mathematical models that emulate some of the observed properties of biological nervous systems and draw on the analogies of adaptive biological learning. The key element of the ANN paradigm is the novel structure of the information processing system. It is composed of a large number of highly interconnected processing elements that are analogous to neurons and are tied together with weighted connections that are analogous to synapses. There are multitudes of different types of ANNs. Some of the more popular include the multilayer perceptron which is generally trained with the backpropagation of error algorithm, learning vector quantization, radial basis function, Hopfield, and Kohonen, to name a few. Some ANNs are classified as feedforward while others are recurrent (i.e., implement feedback) depending on how data is processed through the network. Another way of classifying ANN types is by their method of learning (or training), as some ANNs employ supervised training while others are referred to as unsupervised or self-organizing. Unsupervised algorithms essentially perform clustering of the data into similar groups based on the measured attributes or features serving as inputs to the algorithms. This is analogous to a student who derives the lesson totally on his or her own. ANNs can be implemented in software or in specialized hardware. Learning algorithm and architectureThe neural network used here is the standard feed-forward network, which always passes information in the forward direction. The learning algorithm is the standard backpropagation of error, which minimizes the difference between the actural output of the network and the desired output for patterns in the training set. Training is done on a set of 426 non-homologus protein chains by 7-fold cross-validation. Two feed-forward networks are used. In the first step, the sequence-to-structure network is trained where the occurrence of various residues in a window size of 9 amino acids is correlated with the output of the central residue. The predicted turn/nonturn output from the first net is incorporated along with predicted secondary strcuture information in the second filtering structure-to-structure network and the net is trained to filter the unreasonably isolated residues which are predicted as turns. To generate the neural network architecture and the learning process, the publicly available free simulation package SNNS version 4.2 is used. Click here to download SNNS(v4.2). In both the single sequences and multiple alignment, the network uses a sliding window of 9 amino acids, where a prediction is made for the central residue. With single sequence input, a vector of 21 units, where only one unit is set to 1 for a particular residue and the rest are set to zero, encodes each residue. With multiple alignment profile input, the position specific scoring matrix from PSI-BLAST (Altschul et al., 1997) is used as input to the neural network. The matrix has 21 X M elements, where M is the length of the target sequence. Each residue is encoded by 21 real numbers (rather than by a one and 20 zeros). The notation used to define network topology is 9(21)-10-1 network means it has 3 layers, input layer with window size of 9 residues and 21 input units per window position, a hidden layer with 10 nodes, and an output layer with 1 unit. Total number of units in the input layer is 9 X 21 = 189 units. Also, in second net (structure-to-structure) network, the window size of 9 amino acids is used. 4 units encode each residue; one unit for predicted turn/nonturn by the first network and it is either set to 1 or 0 and remaining 3 units for 3 secondary structure states (helix, strand and coil) predicted by PSIPRED(Jones, 1999) Both the first and the second network have a single output unit, the value of which is 1 for a beta-turn residue and 0 for non-beta-turn residue. The learning process consists of altering the weights of the connections between units in response to a teaching signal that provides information about the correct classification. The difference between the actual output and the desired output is minimized (the sum of square error, SSE). During the testing of network, a cutoff value is set for each network and the output produced by the network is compared with the cutoff value. If the output is greater that the cutoff value, then that residue is taken as beta-turn residue while if it is lower, it is considered as non-beta-turn. For each network, the cutoff value is adjusted that it yields the highest accuracy for that network. |
|
Multiple Alignment
Prediction from a multiple alignment of protein sequences rather than a single sequence has long been recognized as a way to improve prediction accuracy (Cuff and Barton, 1999). During evolution, residues with similar physico-chemical properties are conserved if they are important to the fold or function of the protein. The availability of large families of homologous sequences revolutionised secondary structure prediction. Traditional methods, when applied to a family of proteins rather than a single sequence proved much more accurate at identifying core secondary structure elements. The same approach is used here for the prediction of beta turns. It is a combination of neural network and multiple alignment information. Net is trained on the PSI-BLAST(part of PSIPRED) generated position specific scoring matrices. PSI-BLASTIn PSI-BLAST(Position Specific Iterative Blast)(Altschul et al., 1997), the sequences extracted from a Blast search are aligned and a statistical profile is derived from the multiple alignment. The profile is then used as a query for the next search, and this loop is iterated a number of times that is controled by the user. For more information, Click here. The PSIPRED method has been used for secondary structure prediction. It uses PSI-BLAST to detect distant homologues of a query sequence and generate position specific scoring matrix as part of the prediction process (Jones, 1999), and training is done on these intermediate PSI-BLAST generated position specific scoring matrices as a direct input to the neural network. The matrix has 21 X M elements, where M is the length of the target sequence and each element represents the likelihood of that particular residue substitution at that position in the template. It is a sensistive scoring system, whcih involves the probabilities with which amino acids occur at various positions. |
Performance Measures
|