DataSet:
For the development of effective QSAR model, we have used total 1213 molecules, which were collected from ICANN,2009 (International Conference on Artificial Neural Networks, Eurapion Neural Network society (ENNS) and CADSTAR project), who took the challenge for environmental toxicity prediction (http://www.cadaster.eu/node/67). Data was provided in the form of 644 Training dataset, 449 Known dataset and 120 blind dataset. In this experimental data for the blind set has not been yet previously published and organic molecule for training and known were mainly belonging to the previously published data of Schltz group (see more information at http://www.vet.utk.edu/TETRATOX/). Total 1213 chemical molecules (including Training, known and blind data) were downloaded in mol2 format with pIGC50 values (logarithm of 50% growth inhibitory concentration means log(IGC50-1)) optimized using MOPAC. In this study, dataset was divided in two parts, former set consisted of 1111 randomly chosen compounds as training Dataset for models development and remaining 100 compounds represented the blind dataset. The training dataset was used for five-fold cross-validation training and blind dataset used for checking the applicability of model on blind data (Data available in supplementary dataset) For deriving the structural activity relationship of each molecule, we calculated descriptors using different softwares, V-life, PowerMV and CDK.V-life MDS (Molecular Design Suite) is a workbench for computer aided drug design (CADD) and molecule discovery. Through V-life software, we have calculated ~1002 descriptors including 1D,2D and 3D descriptors. PowerMV a window based software calculates ~6000 descriptors. Another source is Chemistry Development Kit (CDK), a Java based open source library for structural chemo- and bioinformatics projects. by which we have computed 178 descriptors. Feature selection through Weka: In a QSAR study, selection of a preferred set of molecular descriptors is an important step to successfully derive a predictive QSAR model. Initially, the descriptors were selected using CfsubsetEval module with best fit algorithm implemented in weka.For further selection of relevant molecular descriptors F-steeping approach has been employed to remove descriptors irrelevant for the prediction of aqueous toxicity of small chemical molecules in T. pyriformis. Weka Classifiers:
In this study SMOreg algorithm prefom well to predict aqueous toxicity of small chemical molecules in T. pyriformis.Sequential Minimization Optimization for regression (SMOreg) is a new algorithm for training SVM. This implementation globally replaced all missing values and transformed nominal attributes into binary ones. It also normalized all attributes by default. Performance Measures:
Once a regression model was constructed, goodness about the fit and statistical significance was assessed using the statistical parameters outlined below.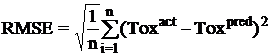
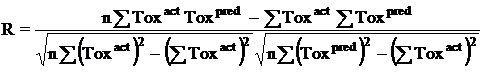
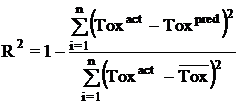 Where n is the size of test set, m is the size of training set, Toxpred is the predicted pIGC50 and Toxact is the actual pIGC50, is the toxicity in test set, RMSE are the root mean squared error between predicted & actual pIGC50 of compounds, R is the Pearson's correlation coefficient between actual and predicted value, R2 (Coefficient of determination) is the statistical parameter for proportion of variability in model. The coefficient of determination is also the arithmetic average of all five folds.
Where n is the size of test set, m is the size of training set, Toxpred is the predicted pIGC50 and Toxact is the actual pIGC50, is the toxicity in test set, RMSE are the root mean squared error between predicted & actual pIGC50 of compounds, R is the Pearson's correlation coefficient between actual and predicted value, R2 (Coefficient of determination) is the statistical parameter for proportion of variability in model. The coefficient of determination is also the arithmetic average of all five folds.
For the development of effective QSAR model, we have used total 1213 molecules, which were collected from ICANN,2009 (International Conference on Artificial Neural Networks, Eurapion Neural Network society (ENNS) and CADSTAR project), who took the challenge for environmental toxicity prediction (http://www.cadaster.eu/node/67). Data was provided in the form of 644 Training dataset, 449 Known dataset and 120 blind dataset. In this experimental data for the blind set has not been yet previously published and organic molecule for training and known were mainly belonging to the previously published data of Schltz group (see more information at http://www.vet.utk.edu/TETRATOX/). Total 1213 chemical molecules (including Training, known and blind data) were downloaded in mol2 format with pIGC50 values (logarithm of 50% growth inhibitory concentration means log(IGC50-1)) optimized using MOPAC. In this study, dataset was divided in two parts, former set consisted of 1111 randomly chosen compounds as training Dataset for models development and remaining 100 compounds represented the blind dataset. The training dataset was used for five-fold cross-validation training and blind dataset used for checking the applicability of model on blind data (Data available in supplementary dataset) For deriving the structural activity relationship of each molecule, we calculated descriptors using different softwares, V-life, PowerMV and CDK.V-life MDS (Molecular Design Suite) is a workbench for computer aided drug design (CADD) and molecule discovery. Through V-life software, we have calculated ~1002 descriptors including 1D,2D and 3D descriptors. PowerMV a window based software calculates ~6000 descriptors. Another source is Chemistry Development Kit (CDK), a Java based open source library for structural chemo- and bioinformatics projects. by which we have computed 178 descriptors. Feature selection through Weka: In a QSAR study, selection of a preferred set of molecular descriptors is an important step to successfully derive a predictive QSAR model. Initially, the descriptors were selected using CfsubsetEval module with best fit algorithm implemented in weka.For further selection of relevant molecular descriptors F-steeping approach has been employed to remove descriptors irrelevant for the prediction of aqueous toxicity of small chemical molecules in T. pyriformis. Weka Classifiers:
In this study SMOreg algorithm prefom well to predict aqueous toxicity of small chemical molecules in T. pyriformis.Sequential Minimization Optimization for regression (SMOreg) is a new algorithm for training SVM. This implementation globally replaced all missing values and transformed nominal attributes into binary ones. It also normalized all attributes by default. Performance Measures:
Once a regression model was constructed, goodness about the fit and statistical significance was assessed using the statistical parameters outlined below.
Where n is the size of test set, m is the size of training set, Toxpred is the predicted pIGC50 and Toxact is the actual pIGC50, is the toxicity in test set, RMSE are the root mean squared error between predicted & actual pIGC50 of compounds, R is the Pearson's correlation coefficient between actual and predicted value, R2 (Coefficient of determination) is the statistical parameter for proportion of variability in model. The coefficient of determination is also the arithmetic average of all five folds.