 |
Algorithm
Dataset Information
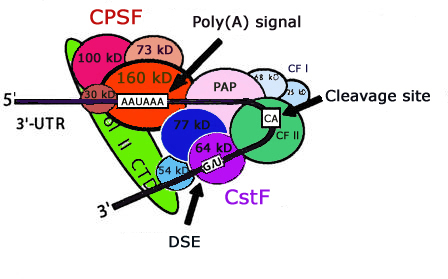
The dataset used in this study to train the SVM was collected from Huiquing, L., Hao, H., Jinyan , L., (2003) Genome informatics 14, 84-93.The positive and negative training data contains 2327 & 2333 sequences respectively and every sequence having 206 nt. long containing AATAAA or (NNTANA) pattern located in the middle of sequence.
Support Vector Machine
Support Vector Machine
Support vector machine (SVM) is the kernel based machine learning method presented by V.N.Vapnik. It has been successfully applied to numerous classification and regression tasks in bioinformatcs. In this study, a freely downloadable package of of SVM, SVM_light, has been used (http://ais.gmd.de/~thorsten/svm_light/). We used RBF kernel to classify the data of PASes and pseudo-PASes. The regulatory parameters c and g of RBF kernal were optimize to 2 and 0.001, respectively.
The SVM_light is used to predict the human polyadenylation signal.The SVM modules were developed by using different nucleotides frequency in each region around Polyadenylation signal (PAS) and combined them..
Evaluation of Performance:-
The 5 fold cross validation technique examined the prediction
quality. In this technique the relevant dataset was partitioned
randomly into 5 equal datasets. The training and testing was carried
out five times, each time onset for testing and other 4 sets for
training. The accuracy of results commonly measured by the quantity
of True Positives (TP), True Negatives (TN), False Positives (FP)
and False Negatives (FN). In the prediction system the sensitivity, specificity and Mathew's correlation co-efficient (MCC) was calculated by following equations.
Sensitivity = TP / (TP+FN),
Specificity = TP / (TP+FP),
MCC = (TP * TN) - (FP*FN) / Ö(TP+FN)* (TP+FP)*(TN+FP)*(TN+FN).
In this method we developed mixed pattern as an input feature by using different nucleotides frequency frequency of 100nt long upstream sequence combined with frequency of 100 nt long downstream sequence relative to Polyadenylation signal (PAS).The maximum MCC achieved using mononucleotide, dinucleotide and trinucleotide and tetranucleotide frequency were 0.51, 0.62, 0.67 and 0.68 respectively.To obtain more information about region specific distinct base elements, we split each 100nt long sequence into two regions and features of each region were combined to develop SVM model.Finally we developed a hybrid method, which combination frequency of dinucleotide, pseudo-dinucleotide and tetranucleotide of each region and achieved maximum MCC of 0.72.
|
|

