|
TAP is an transporter assiociated with MHC class I restricted antigen processing.The TAP is heterodimeric transporter belong to the family of ABC transporter, that uses the energy provided by ATP to transloacte the peptides across the membrane.The transporter is composed of two proteins named TAP-1 and TAP-2. The subset of these transported peptide will bind MHC class I molecules and stabilize them. These MHC-peptide complexes will be translocated on the surface of antigen presenting cells (APCs). The adducts of MHC and Peptide complexes are the ligands for T cell receptors (TCR). These complexes elicit the immune response for clearing various intracellular infections.
How does the TAP complex work is shown in the figure below.
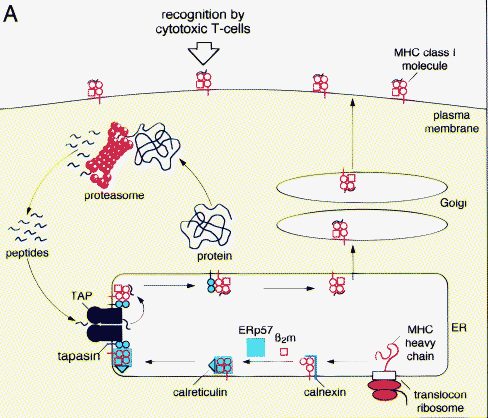
Detailed mechnaism of transport by TAP transporter
Peptide transport by TAP is a multi-step process. In a fast bimolecular association step, the peptide binds to TAP in an ATP-independent manner, followed by a slow isomerization of the TAP complex. It is suggested that this structural reorganization of the molecule triggers ATP hydrolysis and peptide translocation across the membrane. These binding steps primarily determine the selectivity of TAP. The translocation strictly requires hydrolysis of ATP, because non-hydrolyzable ATP analogs do not promote peptide transport. ATP and ADP have similar affinities for TAP; therefore, peptide translocation can be inhibited by ADP.
Peptide binding to TAP transporter
Due to extensive polymorphism of TAP transporter , distinct set of peptides will be translocated to ER. The natrure of these peptides is reflected in the nature of MHC binding peptides The selective transport of the peptides by TAP may modulate or limit the supply of the peptides to HLA class I molecules. Thus, the molecular understanding of the selectivity and specificity of TAP may contribute dramatically in the prediction of the MHC class I restricted T cell epitopes.��The TAP transporter efficently bind and transport the peptides of 8-12 amino acids. It appears TAP binds peptides that are of optimal length or slighly larger then those presented by MHC class I molcules.In spite of length preference the nature of peptides has an influence on peptide selectivity. TAP from the Rat strain RT1a as well as human TAP translocate peptides with broad specificity (hydrophobic or basic amino acids at COOH terminus), whereas TAP from rat strain RT1u and TAP prefers the peptides with hydrophobic COOH termini. According to another observation TAP favours strongly hydrophobic residues in position 3 (P3) and hydrophobic and charged residues in P2, whereas aromatic and acidic residues in P1. Van Endert and Coworkers also observed that proline in position 1 and 2 have very deterious effect on binding.The TAP specificity obtained by the Peptide specificity for the TAP transporter as determined by combinatorial peptide libraries
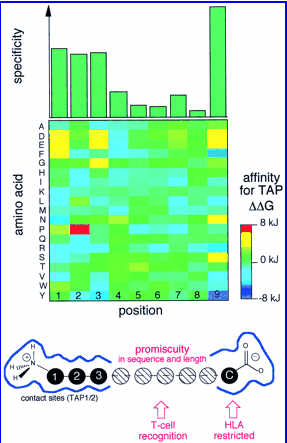
The figure has been obtained from Lankat-Buttgereit et al., 2002. Top panel: substrate specificity for TAP. The first three NH2-terminal amino acids and the last COOH-terminal amino acid contribute significantly to the stabilization of peptide binding to TAP. Middle panel: favored amino acids at the individual positions with negative Delta Delta G values (favored residues) are shown in blue, and positive Delta Delta G values (disfavored residues) are in red. For example, for the first position the amino acids K, N, and R are favored, and D, E, and F are disfavored. Bottom panel: a model of the substrate-binding pocket of TAP.
Why computaional method is required for prediction of TAP binding affinity of peptides?
The wet Lab testing of the peptides deived from the proteins is experimantally laborious and economically expensive.The Prediction methods based on the specificity of TAP transporter will complement the wet lab experiments and speed up the knowledge discoveries.on the basis of this two computational algorithms were dedeveloped in past. The algorithms are based on the machine learning technique(ANN).
|