About Propred1
Source of Weight Matrices used in Propred1
Algorithm for Prediction of MHC Binders
Prediction of Proteasome Cleavage Sites
Simultaneous Prediction of MHC binders and Proteasome
Cleavage sites
Selection of Parameters
Performance of Propred1
A
Case Study
Presentation of Results
Limitations of Propred1
An Example Submission Form
Go Top
About Propred1
ProPred1 is an on-line web tool
for the prediction of peptide binding to MHC class-I alleles. This is
a matrix based method that allows the prediction of MHC binding sites in
an antigenic sequence for 47 MHC class-I alleles. The matrices used in
ProPred1 have been obtained from BIMAS server and from the literature.
ProPred1 also allows the prediction of the standard proteasome and immunoproteasome
cleavage sites in an antigenic sequence. This server has implemented the
matrices described by Toes et al., 2001 for the identification
of proteasome (standard/constitutive proteasome and immunoproteasome.)
cleavage site in an antigenic sequence. It allows filtering of MHC binders,
who have cleavage site at C terminus. Recently, Kessler et
al., 2001 and Ayyoub et al., 2002 have demonstrated that
most of MHC binders having Proteasome cleavage site at their C terminal
have high potency to become T cell epitopes. In brief, the server assists
users in identification of promiscuous potential T-cell epitopes in an
antigenic sequence. These epitopes can serve as suitable vaccine candidates.
The server represents MHC binding regions and proteasome cleavage sites
in an antigenic sequence in user-friendly formats by presenting the output
in graphical or text format. These display formats help the user in easy
detection of the promiscuous MHC binding regions in their query sequence.
Propred1 has been installed
on a Sun Server (420E) under UNIX (Solaris 7) environment and launched
using Apache web server. Most of the programs including common gateway
interface (CGI) scripts are written in PERL The protein sequences can be
submitted to the ProPred1 by cut-and-paste technique or by directly uploading
a sequence file. The server uses ReadSeq (developed by Dr. Don Gilbert)
to parse the input sequence, therefore it can accept most of the commonly
used sequence formats. The server allows user to select the threshold for
their prediction. The threshold plays a vital role in determining the stringency
of prediction. Lower the threshold, higher is the stringency of prediction
i.e. lower rate of false positives and higher rate of false negatives in
the prediction. In contrast, a higher threshold value (low stringency)
corresponds to a higher rate of false positives and a lower rate of false
negatives.
Go Top
Source of Weight Matrices used
in Propred1
Matrices for the
Prediction of MHC Binders: The matrices used in Propred1 for the
prediction of MHC binders for 7 MHC Class I alleles were mostly from BIMAS
server. Few matrices were obtained from published literature. Following
table provides complete information about sources of matrices (See Matrices for detail).
|
Name of MHC allele
|
Reference
|
Comment
|
|
HLA-A2.1
|
Ruppert et al., 1993
|
Addition Matrix
|
|
HLA-B*0702
|
Sidney et al., 1996
|
Addition Matrix
|
|
HLA-B51
|
Sidney et al., 1996
|
Addition Matrix
|
|
HLA-B*5301
|
Sidney et al., 1996
|
Addition Matrix
|
|
HLA-B*5401
|
Sidney et al., 1996
|
Addition Matrix
|
|
All other MHC
alleles
|
Unpublished
|
Multiplication
Matrices (BIMAS Server)
|
Matrices for the
Prediction of Cleavage Sites: The weight matrices
for both standard proteasome and immunoproteasome were derived from Table
1 & 2 in the paper of Toes et al., 2001. Each value of these tables
were divided by 1000 in order to rationalize the score (See Matrices for detail).
Go Top
Algorithm for the Prediction
of MHC Binders
The ProPred-1 utilizes matrix
data in a linear prediction model where contribution of each amino acid
is summed up multiplied depending which type of matrix is used for prediction.
The peptides having scores more than a defined (threshold score) are
assign as binders. Following is the brief description of algorithms.
Computation of Score
Multiplication
Matrices: The most of matrices in Propred1 is multiplication type where
the score is calculated by multiplying scores of each position. For example,
score of peptide ‘PACDPGRAA” can be calculated by following equation.
Score = P(1) ´ A(2) ´ C(3) ´ D(4) ´ P(5) ´ G(6) ´ R(7) ´ A(8) ´ A(9) (1)
Where P(1)
is score of P at position 1.
Addition Matrices: The matrices obtained from the literature are “Addition
Matrices”, where score is calculated by summing the scores of each position.
For example, score for above peptide “PACDPGRAA” is calculated as follows.
Score = P(1) + A(2) + C(3) +
D(4) + P(5) + G(6) + R(7) + A(8) + A(9) (2)
Calibration of Threshold Score
for each Allele
One of the crucial steps
in matrix based methods to adjust the cut-off score called threshold score, as we obtained these matrices
from various sources so its not clear what should be threshold score.
The number does not give any sense so we adjust the score in such a way
that user can select threshold score in term of percent such as 3% , 4%
etc. For example 4% threshold score means that there is 4% chance that
your predicted binder is random peptide. We follow the following steps
in order to calculate threshold score for each allele/matrix.
i)
All proteins were obtained from SWISSPROT
databases for creating the overlapping peptides of length nine. For example,
a protein of length n will have (n+1 – 9)
overlapping peptides.
ii)
The score of all natural 9-mer peptides have
been calculated using weight matrix of that allele. These peptides have
been sorted on the basis of score in descending order and top 1 % natural
peptides have been extracted. The minimum score that we called threshold
score was determined from these selected peptides.
Similarly, threshold scores at 2%, 3% … 10% were calculated.
iii)
Step 1 and 2 were repeated for each MHC allele
in order to calculate threshold score at different percent for each allele
used in ProPred1.
Identification of MHC binders
In order to identify
the MHC binder in an antigen sequence, first Propred1 generate the overlapping
9-mer peptides. In next step, the score of these 9-mer peptides are calculated
using quantitative matrix of selected MHC alleles. Finally, all peptides
having score greater than selected threshold score (e.g. at 4%) are
considered as predicted binders for selected MHC allele. Predicted binders
are presented on antigen sequence by different color or along the primary
sequence.
Go Top
Prediction of Proteasome Cleavage
Sites
The prediction
proteasome cleavage site is based on Toes et al., 2001 work. We derived
the matrices for standard proteasome from Table 1 of Toe et al., 2001.
The derived matrix is an “Addition Matrix” where score of a peptide is
calculated by summing the score at each position (See ‘Computation of score’
subsection of section ‘Prediction of MHC binders’ for detail). Similarly,
procedure has been adopted for deriving the matrix for immunoproteasome,
from the Table 2 of Toes et al., 2001. The major difference
between proteasome matrices and MHC matrices is that proteasome matrices
consider the peptide of length twelve instead of nine. The cutting site
is at the center of 12-mer peptide.
Calibration of Threshold Score
Threshold score
for proteasome prediction was computed, in order to provide the confidence
to the users. The threshold scores for standard proteasome and immunoproteasome
have been calculated at different percent by using the approach described
above for calculation of threshold score for MHC alleles. The calculation
of threshold score of proteasome matrices requires the 12-mer overlapping
peptides. The matrices and cutoff scores at different threshold 1%, 2%,
… 10% are available at URL http://webs.iiitd.edu.in/raghava/propred1/matrices/matrix.html
.
Identification of Cleavage Site
In order to predict proteasome
cleavage sites in an antigenic sequence. The overlapping 12-mer peptides
were generated for antigenic sequence and score of these peptides were
calculated using weight matrix of proteasome. In next step, all peptides
having score greater than selected threshold score (e.g. at 4%) are considered
as peptides having proteasome cleavage site. The center positions of these
peptides (6-position left and 6 position right) are considered as predicted
proteasome cleavage site. Similar approach has been utilized for prediction
of peptides having immunoproteasome cleavage site. .
Go Top
Simultaneous Prediction of
MHC Binders and Proteasome Sites
One of the powerful feature
of Propred1 is that it allows prediction of MHC binders for various alleles
and proteasome cleavage site, simultaneously. This is based on observations
of previous studies where it has been demonstrated that most of MHC binders
having Proteasome cleavage site at their C terminus have high potency
to become T cell epitopes (Kessler et al., 2001 and Ayyoub
et al., 2002). The predicted MHC binders are filtered
based on prediction of proteasome cleavage sites in an antigenic sequence.
Firstly, the server computes the predicted MHC binders and their C terminus
position for a selected MHC allele in an antigenic sequence. Secondly
server predicts the cleavage sites of proteasome (standard proteasome,
immunoproteasome or both) in an antigenic sequence at given threshold
(e.g. at 4%). Finally, all predicted MHC binding peptides whose C terminal
position coincides with proteasomes cleavage sites were filtered. These
peptides were also called predicted potential T-cell epitopes. In other
words, we removed the MHC binders from list which does not have proteasome
site at C terminous.
Go Top
Selection
of Parameters
Selection
of Alleles: Propred1 allows user o select the any allele or combination
of alleles or all alleles (total 47) of MHC class I. The server will
predict the MHC binders of these selected alleles in an antigen/protein
sequence.
Threshold
Score for the Prediction of MHC Binders: The server have default
threshold 4%, user may select their own threshold depending on need.
we observed that most of the alleles have sensitivity and specificity
nearly same at 4% so we set it as default threshold. This is a critical
parameter user should vary it according to requirement, for example if
user is interested to detect all possible binders of alleles than user
should select threshold like 8% or 9% etc. (in this case coverage will
be high but probability of prediction will be very poor), if user is only
interested in top binders with high confidence than user should select
threshold like 1% or 2 % (in this case probability of correct prediction
will be high but coverage will be poor).
Type
of Display: Propred1 allow user to display their result in four formats;
i) HTML 1; ii) HTML-II, Graphical and Tabular format. For detail see the section ‘Presentation of Result’.
Proteasome
Filters: The user can filter their MHC binders who have standard
proteasome of immunoproteasome cleavage sites at C terminus. By default
its off, user can ‘ON’ these filters to see which of their MHC binders
have cleavage site at C-terminus.
Threshold of Proteasome filter: In case user is interested
to use proteasome filters than they should also select cut-off threshold
which is 5 % by default. User can select threshold suitable to their
requirement (See ‘Threshold Score for the Prediction of MHC Binders’
sub section for detail).
Go
Top
Performance
of Propred1
What is the performance
of server for various alleles ? or How much I can rely on prediction
? . This is one of the obvious questions in users mind when they use any
prediction server.
Percent
Coverage: We calculated the percent coverage (percent of binders
correctly predicted as binder) for each allele for which sufficient amount
of data was available. The data of binders and
non binders corresponding to each MHC alleles has been extracted from
MHCBN database (http://webs.iiitd.edu.in/raghava/mhcbn/ Bhasin et al., 2002). The number of binders
varies from 20 to 1200. Following table shows the result at default threshold
score 4% (score at which sensitivity and specificity are nearly the same).
The percent coverage has been calculated from predicted results.
|
MHC Alleles (Total binder, % Coverage)
|
|
HLA-A*0201(1221,
75%)
|
H2-Db (189,
74%)
|
HLA-B*0702(79,
92%)
|
|
HLA-A*0205(28,
61%)
|
H2-Dd (89,
74%)
|
HLA-B*2705(145,
98%)
|
|
HLA-A*1101(116,
80%)
|
H2-Kb (116,
78%)
|
HLA-B*3501(254,
84%)
|
|
HLA-A*3101(33,
70%)
|
H2-Kd (277,
83%)
|
HLA-B*5101(51,
92%)
|
|
HLA-A1
(128, 77%)
|
H2-Kk (28,
86%)
|
HLA-B*5102(33,
94%)
|
|
HLA-A2
(976, 69%)
|
H2-Ld (113,
60%)
|
HLA-B*5103(30,
97%)
|
|
HLA-A2.1
(77, 64%)
|
HLA-B*5401(60,
100%)
|
HLA-B8
(130, 75%)
|
|
HLA-A24
(60, 70%)
|
HLA-B61
(22, 95%)
|
HLA-B62
(29, 55%)
|
|
HLA-A3
(191, 64%)
|
HLA-B14
(81, 75%)
|
HLA-Cw*0401(20,80%)
|
|
HLA-B7
(134, 81%)
|
HLA-B*5301
(64, 95%)
|
|
These results
clearly indicate that in most of the cases percent coverage is more than
80% which is reasonably good. Almost all alleles showed reasonable percent
coverage, which means threshold criteria and matrices used in ProPred1
are beneficial for experimental Professors.
Comprehensive Evaluation of
ProPred1: Nonetheless,
the percent coverage is a useful measure to evaluate the ability of method
for the identification of binders from a given sequence, but it does not
provide any information about predicted false positive binders or accuracy
of prediction etc. Thus we also perform comprehensive evaluation of Propred1,
where following three commonly used parameters were used to measure the
performance.
 [1]
[1]
 [2]
[2]
 [3]
[3]
The correlation coefficient
(CC) is a rigorous parameter to measure the performance of a method,
which is commonly used in other fields of the science (e.g. secondary
structure prediction). The CC can be defined as:
 [4]
[4]
Where TP and TN are correctly
predicted binders and non binders respectively. FP and FN are wrongly
predicted binders and non binders respectively.
We compute all
the above parameters for ProPred1 for its comprehensive evaluation. In order to evaluate a method one need sufficient data
of experimentally proven MHC binders and non binders. Unfortunately,
most of the alleles have very limited number of binders and . non bindersThus,
the comprehensive evaluation of ProPred1 was performed only for two alleles
(HLA-A*0201 & H2-Kb) for which sufficient number of binders and non
binders binders were available. The peptides for allele HLA-A*0201 (1220
binders & 56 nonon binders and H2-Kb (300 binders & 200 bnon bindersibinders
werebtained from MHCBN database (Bhasin et al., 2002). The performance
of ProPred1 for these two MHC alleles at different percent threshold has
been shown in following table
|
Thres-
holds
|
HLA-A*0201
|
H2-Kb
|
|
Sensitivity
(%)
|
Specificity
(%)
|
Accuracy
(%)
|
Correlation
coefficient
|
Sensitivity
(%)
|
Specificity
(%)
|
Accuracy
(%)
|
Correlation
coefficient
|
|
1%
|
36
|
98
|
38
|
0.1314
|
68
|
88
|
70
|
0.372
|
|
2%
|
57
|
93
|
58
|
0.1854
|
73
|
81
|
74
|
0.3775
|
|
3%
|
66
|
80
|
67
|
0.1783
|
78
|
81
|
78
|
0.4209
|
|
4%
|
75
|
78
|
75
|
0.2179
|
78
|
69
|
77
|
0.3367
|
|
5%
|
81
|
67
|
80
|
0.2151
|
82
|
62
|
80
|
0.3418
|
Go Top
A Case Study
The purpose of development of ProPred1 is to
effectively reduce number of wet lab experiments involved in the identification
of potential T cell epitopes or suitable vaccine candidates. In order
to demonstrate the usefulness of Propred1 in real life, we applied Propred1
on an antigen which has been extensively studies and whose MHC binders
and T cell epitopes have been identified experimentally. Recently, Kessler et al., 2001 have
experimentally determined the MHC binders and T cell epitopes from tumor
associated antigenic protein, PRAME. We analyzed the performance of ProPred1
in the identification of experimentally proven MHC binders and T cell
epitopes of PRAME. The sequence of PRAME antigenic protein was obtained
from SWISSPROT database.
MHC Binder:
Kessler et al., 2001 tested 128 peptides and identified 19 as high-affinity
binders and 27 intermediate-affinity binders. ProPred1 was used to predict
these MHC 128 peptides of PRAME at various thresholds. Following table
shows the performance of Propred1.
|
Threshold (%)
|
Correctly predicted high-affinity binders (out of 19)
|
Correctly predicted intermediate-affinity binders (out of 27)
|
|
1.0
|
4 (21%)
|
1 (4%)
|
|
2.0
|
9 (47%)
|
6 (22%)
|
|
3.0
|
10 (53%)
|
14 (52%)
|
|
4.0
|
11 (58%)
|
15 (56%)
|
|
5.0
|
12 (63
%)
|
21 (77%)
|
|
6.0
|
13 (68%)
|
22 (81%)
|
|
7.0
|
13 (68%)
|
23 (85%)
|
|
8.0
|
15 (79%)
|
24 (89%)
|
|
9.0
|
18 (95%)
|
26 (96%)
|
|
10.0
|
19 (100%)
|
27 (100%)
|
As shown in Table, number
of correctly predicted binders (intermediate/high affinity) depend on
percent threshold. The ProPred1 predicted all binders correctly at 10%
threshold. This clearly indicate that server has capability to predict
the binders. The performance of ProPred1 was significant even at 4% threshold
(default threshold). The default threshold is that threshold at which
sensitivity and specificity of a method is nearly the same.
Potential T-cell
epitopes: It has been demonstrated experimentally that MHC binders
having Proteasome cleavage site at C-terminus are mostly responsible for
the activation of cytotoxic T lymphocytes (CTLs). Kessler et al., 2001
experimentally identified four regions having HLA-A*201 restricted T cell
epitopes. We tested these regions using ProPred1 server. Firstly, binding
regions were predicted at default threshold (4%) in protein PRAME. Secondly,
all proteasomes sites were predicted at various thresholds. Finally, predicted
binders having proteasomes cleavage site at C-terminus were identified. The number of peptides predicted by above falls in regions
identified as T cell epitopes by Kessler et al., 2001, is shown in following Table. Propred1 was tested on 4 regions of
PRAME (A: 90-116; B:133-159; C: 290-316; D: 415-441) which were identified
as T cell epitopes by Kessler et al., (2001). The column 2 shows the
number of predicted peptides and regions (in bracket), which agree with
the experimentally identified epitopes.
|
Name of
filter
|
Correctly
predicted T cell epitopes in protein PRAME at different thresholds (out
of 4)
|
|
2%
|
3%
|
5%
|
7%
|
|
Standard
Proteasome
|
0
|
1 (A)
|
1 (A)
|
2 (A,D)
|
|
Immunoproteasome
|
2 (A,D)
|
2 (A,D)
|
3 (A,C,D)
|
3 (A,C,D)
|
|
Immunoproteasome or
Standard Proteasome
|
2 (A,D)
|
2 (A,D)
|
3 (A,C,D)
|
3 (A,C,D)
|
Table shows
the regions where predicted T cell epitopes by ProPred1 and experimentally
identified T-cell epitopes matched. It was observed that in the presence
of standard proteasome filter at 7%, the server was able to predict the
50% of binding regions that are in agreement with experimentally proven
binding regions as demonstrated by Kessler et al., 2001.
Similarly, it has been observed that at 5% of threshold of immunoproteasome
filter, the server was able to identify 75% of experimentally determined
binding regions. The server was able to predict 75% of binding regions
in simultaneous presence of either standard proteasome or immunoproteasome
filters at 5% threshold. Hence, all the analysis clearly indicate that
it is worth using ProPred1 for the identification of MHC binding regions
having Proteasome cleavage site at their C terminus or potential T cell
epitopes.
Go Top
Presentation of Results
One of the important
aspects of MHC prediction is the representation of binding peptides found
within the antigenic sequence. This can be achieved by developing a powerful
interface of prediction methods. The ProPred1 provides three major options
to visualize results in user-friendly formats, including most popular
tabular format. Following is the brief description of these options.
Graphical Display: The graphical output represents the quantitative estimation
of MHC binding propensity of the antigenic sequence. The server represents
results in graphical format (X-Y Plot), where amino acid sequence is
shown along the sequence and peptide score is shown along the . Y- axisThe
images are generated in GIF format using the GDPlot library (developed
by Lincoln D. Stein). Each binder is represented as a peak crossing the
dashed threshold line in the image. Besides this, the server also plots
the threshold profile (threshold versus binding peptides). This profile
assists experienced users in selecting the threshold for locating the
promiscuous regions in antigenic sequence. It allows user to locate the
promiscuous regions in the query sequence by looking at the peaks in graphs
for different MHC alleles. Each binder is represented as a peak crossing
the dashed threshold line. Following is the example of graphics output.
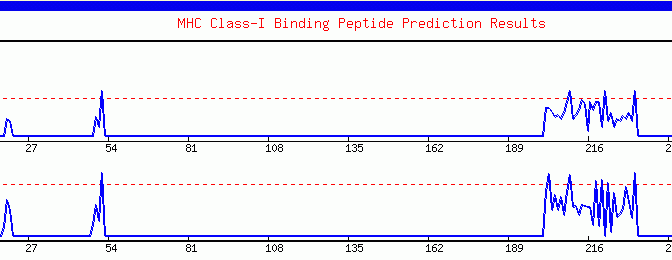
Graphical
output generated by the ProPred-1. The peaks (starting from ~200th amino
acid) crossing the red threshold line are the predicted binders.
Text or HTML
Format: This option of server presents the MHC binders within antigenic
sequence in text or HTML format. It has two sub-options. The first sub
options displays the predicted MHC binders in separate lines along the
antigen sequence. This option uses the separate lines for representing all
the predicted overlapping binders within the sequence. This suboptions is
very useful for viewing the predicted overlapping binders. In this option (HTML-I), the overlapping regions
are presented on separate lines making it easier to detect the overlaps.

The HTML-I output: The prediction is made at 7% threshold
for explaining the output. The peptide frames "RTFEREYRT", "FEREYRTRL",
and "REYRTRLKT" are represented more expressively than the simple "RTFEREYRTRLKT"
string.
The second osuboption of
the server represents predicted binder by different color i.e. blue. The
first position of each binder is shown by red color so that user can easily
distinguish the overlapping peptides. This option (HTML-II)
is similar to that used by TEPITOPE and ProPred.

The peptide
"YLESQLEEL" is predicted to bind to five alleles. The main advantage
of this display is that it allows easy way to locate promiscuous regions
in sequence (a region that can bind to number of MHC alleles). For example
peptide "QQRTVLEGRLEQLRTFEREYRTRLKTYLESQLEEL" binds to ten MHC alleles
out of eleven MHC alleles. Though useful, this option is less expressive
in presenting the overlapping binding regions.
Tabular Format:
This is the most widely used option for the display of results in most
of the web servers of MHC prediction. This option displays the peptides
sorted in descending order of their score. The server creates a separate
table corresponding to each selected allele. Following
is the example output of Propred1 for Table Format.
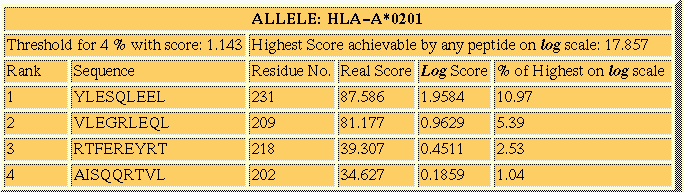
Go Top
Limitations of ProPred1
All the matrices used in server
were obtained from various servers and from the literature. The base
of selection of matrices is on its availability from single source and
not on the performance. Thus, it is not necessary that we are using best
matrix for an allele if more than one matrix is available in the literature.
In this server only 9mer peptide length are predicted not 8mer or 10mer.
Thus it is possible that ProPred1 may miss potential 8mer and 10mer binders.
The matrices for predicting ProPred1 were obtained from the paper of
Toes et al. (2001), where their values were obtained for enolase-I protein.
We have used these values for all predictions, it is not necessary that
this generalization will work for all the proteins.
Go Top
|