The data for four subfamilies of nuclear receptors was obtained from nucleaRDB database available at http://www.receptors.org/NR/. All the entries, which are not marked as fragments, are extracted from database by text parsing method. The initial dataset have 577 sequences belonging to four subfamilies of nuclear receptors. Redundancy was reduced such that none had >=90% sequence identity with any other sequence is data set using PROSET software. The final dataset have 282 sequences belonging to various sub-families of nuclear receptors as shownbelow

Statergy for Prediction Method development:-The method was designed on the basis of support vector machines. SVM was implemented using freely downloadable software SVM_light.This software enables the user to define a number of parameters, as well as select a choice of inbuilt kernel functions including a radial basis function (RBF), or a polynomial kernel (of given degree). Here, all the parameters of kernel are kept constant except regulatory parameter C. The prediction of subfamilies of nuclear receptors is a multiclass classification problem. In this case the number of subfamilies of nuclear receptors are equal to 4. To handle this multiclass situation, we have designed a series of binary SVMs. For N class classification, N SVMs are constructed. The ith SVM will be trained with all samples of ith subfamily with positive label and all the samples of all other subfamilies as negative label. The SVM trained in this way are referred as 1-v-r SVMs . In such classification each of unknown protein will achieve four scores. An unknown protein will be classified into the sub families that correspond to the 1-v-r SVM with highest output score.
Evaluation of method performance:-
The performance of composition as well as dipeptide composition based modules developed in this method is evaluated using 5-fold cross-validation. In 5-fold cross validation dataset was randomly divided into five equal size sets. The training and testing of every module was carried five times, each time using one distinct set for testing and remaining four sets for training. The performance of each module is assessed by calculating the accuracy and Matthew's correlation coefficient (MCC).The formula's of acuuracy and MCC calculation are shown below.

The performence of both amino acid composition as well as dipeptide composition based method is shown below.The overall accuracy of amino acid composition and dipeptide composition based classifiers are 82.6% and 97.5% respectively. This proves that different subfamilies of nuclear receptors are predictable with considerable accuracy by using amino acid or dipeptide composition.

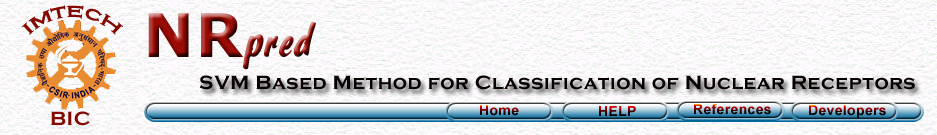
To further bring confidence to user we have calculated reliability index of prediction. The RI provides information about certinity of prediction results. The expected accuracy at different RI values is shown below in form of curve.In case of amino acid composition based classifiers the expected accuracy of sequences with RI=5 is 100%. In whole dataset 32.2% of sequences have RI=5.he dipeptide composition based classifier predicted 84.2% sequences with RI >= 3 and of these sequences are nearly 100% correctly predicted.
Detailed Help:-
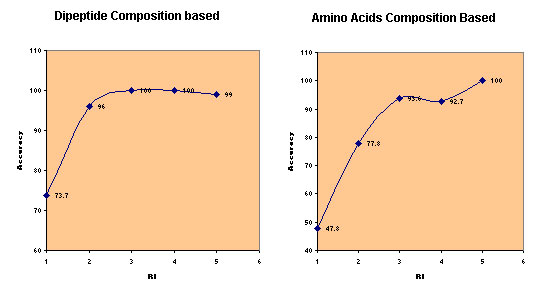
Query Sequence:-The server provides two options for submitting the query sequence. The user can paste plain sequence in the provided inbox.The server also has the facility for uploading the local sequence files. Amino acid sequences must be entered in the one-letter code.All the non standard characters will be ignored from the sequence.A sample of submission form with lebeled fields is shown below.
Sequence Format:-The server can accept both the formatted or unformatted raw antigenic sequences.The server uses ReadSeq routine to parse the input.The user should choose wether the sequence uploaded or pasted is plain or formatted before running prediction.The results of the prediction will be wrong if the format choosen is wrong.
Prediction Approaches
The method allow the prediction on the basis of two different appraoches.
- Composition of amino acids:-
A SVM was developed on the basis of composition amino acids of protein. The SVM was provided with a 20 dimensional vector. The amino acid composition is fraction of each amino acid in a protein.The performence of the method is evalvuated using five fold cross-validation.
- Dipeptide composition
A SVM was developed on the basis of composition of dipeptides of protein sequence. This will give a fixed pattern length of 400. This representation encompasses the information about amino acid composition along local order of amino acid.
Prediction Results
The prediction results are presented in very user friendly format. The results are consist of mainly two parts.
- Summary of query sequence:-
This part provides the information about the submitted sequence like the sequence, length of sequence and date of scanning. This part also provides the information about the choosen prediction approach.
- Prediction Result:-
This part provides information about the final predicted type of amine receptor. It also provides information about reliability of prediction (in form of reliability index) and expected accuracy.A sample of prediction are shown below
Supplementary Material