 |  |
| MMBPred Help Document | |
 |  |
| |
| MMBPred Help Document | |
| |
| |
| Introduction to server | |
| |
The sever will predict promiscuous MHC class-1 binders having mutation at one, two or three position's in each predicted peptide.The promiscuous MHC binders can bind to several HLA alleles. These peptides are most suitable for subunit vaccine development because with single epitope, the immune response can be generated in large population.The mutation in the peptide may increase its binding spectrum of peptide frame.The prediction of the promiscuous MHC binding peptide is crucial due to so much MHC polymorphism. The prediction of the promiscuous MHC binding peptide will increase effectivness of vaccine in different genetic groups.The server will predict mutated binders for 46 MHC class-1 alleles. |
| |
| Prediction Algorithm | |
| |
This server utilizes matrix data in a linear prediction model, in which peptide conformation is entirely neglected. The only parameters describing peptide binding are peptide frames,effect of peptide side chain on binding and peptide length.The complete detail of algorithm can be obtained from MMBPred Algorithm page. |
| |
| Instructions for prediction | |
| |
Step I: Type the following URL address in your web browser
http://webs.iiitd.edu.in/raghava/mmbpred Step II: Select one of the following to display the page having prediction form.
This tool will predict the more immunogenic peptides based on the fact that high affinity binders are more immunogenic. The amino acids of the native peptide frame are replaced by the amino acid which best fit at specific position.The change of amino acid is performed at user defind position. This type of page will appear |
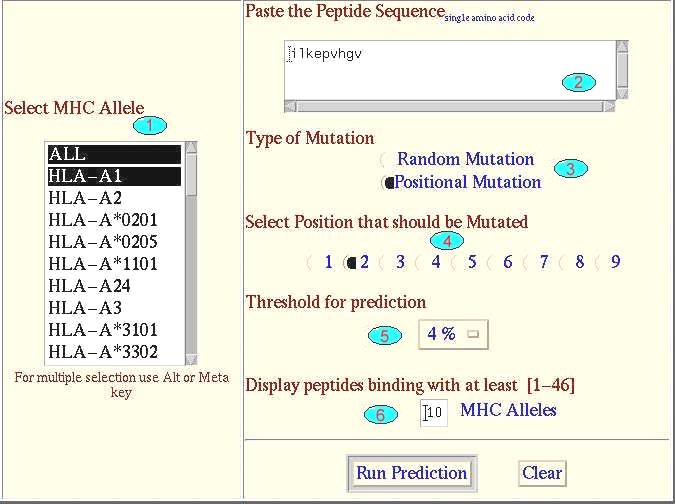
| Length of input sequence | 9 |
| Date of sequence scanning | Fri May 24 17:33:11 2002 |
| Threshold setting | 4% |
| Number of alleles | 46 |
| Peptide binding with at least | 10 MHC allele |
| Position for Mutation[in each Nonamer] | 2 |
