Types of Prediction Methods
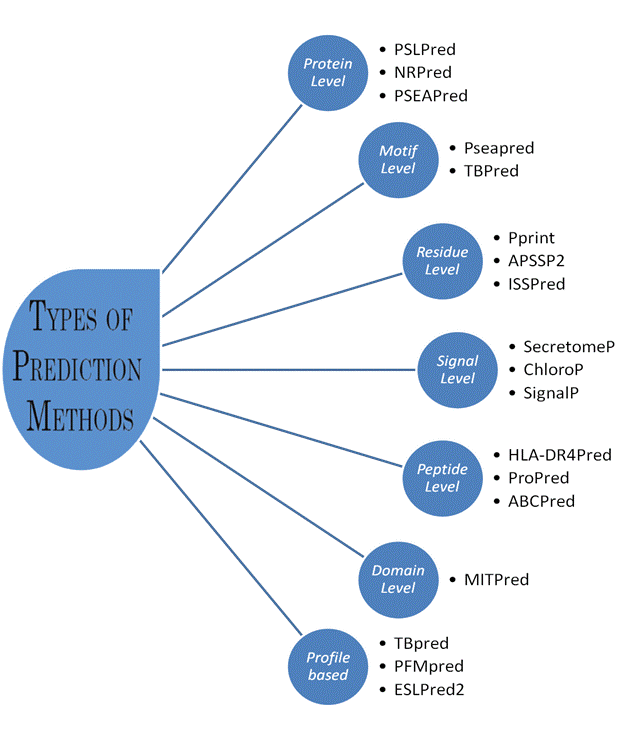
1. Prediction at Protein Level:
These methods are developed to predict overall function of charactestics of proteins. In these methods we used complet protein as input. Following are few examples
1.1 Subcellular level prediction:
The cellular localization of a protein is one of the most fundamental properties of any protein due to cellular division of labour. The correct prediction of subcellular location can be a major breakthrough for functional prediction, since to perform a function, protein must be located in their native location, such as nucleus or mitochondria or outside the cell in case of secretory proteins. The native subcellular localization of a protein is one of the indicators of protein function.
Over the years numbers of methods have been developed for the prediction of subcellular localization in prokaryotes as well as eukaryotes. Existing subcellular localization methods can be divided into various categories:
- Similarity search based techniques: query sequence is searched against experimentally annotated proteins.
Limitation: fail to predict new/novel proteins, if query protein does not have similarity with known proteins.
- Signal sequence based techniques: number of methods fall under this category in which leader sequence or sorting sequence present on protein itself is used for prediction. E.g. TargetP, PSORTb, SignalP
- Sequence composition based techniques: number of methods has been developed so far based on the sequence composition. e.g. SubLoc, NNPSL
- Organism specific and location specific subcellular localization prediction: Organism specific approach is more useful than generalised approach.
Methods
Several computational tools for predicting the subcellular localization of a protein are publicly available, a few of which are listed below:
|
Methods |
Techniques Used |
|
PSLpred |
Composition based+SVM+PSI-BLAST |
|
NRpred |
Composition based |
|
GPCRpred |
Composition based |
|
ESLpred |
SVM+Dipeptide Composition+PSI-BLAST |
|
SRTpred |
Composition based+physic-chemical properties+PSI-BLAST |
|
Cytopred |
SVM+PSI-BLAST hybrid approach |
|
PSEApred |
SVM |
|
PFMpred |
SVM |
|
HSLpred |
Composition based+SVM+PSI-BLAST |
|
NNPSL |
Neural Network |
Relevance
Determining subcellular localization is important for understanding protein function and is a critical step in genome annotation. Knowledge of the subcellular localization of a protein can significantly improve target identification during the drug discovery process. For example, secreted proteins and plasma membrane proteins are easily accessible by drug molecules due to their localization in the extracellular space or on the cell surface.
Subcellular localization prediction allows researchers to make inferences regarding a protein's function, to annotate genomes, to design proteomics experiments and—particularly in the case of bacterial pathogen proteins—to identify potential diagnostic, drug and vaccine targets.
1.2 Class level prediction:
1.2.1 Classification of proteins
GPCRsclass: classification of amine type of G-protein-coupled receptors
1.2.2 Nucleotide binding protein prediction:
Most of the functions of DNA/RNA are performed through interaction with proteins. Prediction of DNA/RNA binding Proteins can be categorised into 2 categories:
Structure based methods: structure based methods can’t be used in high throughput annotation, as they require the structure of a protein for the prediction
Sequence based methods: Only couple of sequence based prediction methods have been developed so far. These methods are based on pseudo-amino acid composition, amino acid composition, composition of physico-chemical properties and Support Vector Machine (SVM).
METHODS:
DISIS: predicts DNA binding sites directly from amino acid sequence
DBS-Pred: predict DNA-binding proteins using amino acid composition
1.3 Family level prediction
Computational prediction and classification of GPCRs can supply significant information for the development of novel drugs in pharmaceutical industry.
GPCRpred: An SVM Based Method for Prediction of families and subfamilies of G-protein coupled receptors
GSTPred: prediction of GST proteins
GPCRsIdentifier: a corresponding stand-alone executable program for GPCR identification and classification.
1.4 Structure class of proteins
Proclass: predict the class of protein from its amino acid sequence
TBBpred: predicts the transmembrane Beta barrel regions in a given protein sequence
- Prediction at Residue level:
2.1 Prediction of Nucleotide binding residues:
Structural and physical properties of DNA provide important constraints on the binding sites formed on surfaces of DNA-binding proteins. Characteristics of such binding sites may be used for predicting DNA-binding sites from the structural and even sequence properties of unbound proteins. This approach has been successfully implemented for predicting the protein-protein interface. Here, this approach is adopted for predicting DNA-binding sites in DNA-binding proteins. First attempt to use sequence and evolutionary features to predict DNA-binding sites in proteins were made by Ahmad et al. (2004) and Ahmad and Sarai (2005). Some methods use structural information to predict DNA-binding sites and therefore require a 3-dimensional structure of the protein, while others use only sequence information and do not require protein structure in order to make a prediction. Structure- and sequence-based prediction of DNA-binding residues in DNA-binding proteins can be performed on several web servers listed below:
- Pprint (Prediction of Protein RNA- Interaction): is a web-server for predicting RNA-binding residues of a protein. The prediction is done by SVM model trained on PSSM profile generated by PSI-BLAST search of 'nr' protein database.
2.2 Post-translational modifications of proteins
ISSPred: Intein Splice Site Prediction
DictyOGlyc: O-(alpha)-GlcNAc glycosylation sites (trained on Dictyostelium discoideum proteins)
NetAcet: N-terminal acetylation in eukaryotic proteins
NetCGlyc: C-mannosylation sites in mammalian proteins
NetCorona: Coronavirus 3C-like proteinase cleavage sites in proteins
NetNGlyc: N-linked glycosylation sites in human proteins
NetOGlyc: O-GalNAc (mucin type) glycosylation sites in mammalian proteins
NetPhos: Generic phosphorylation sites in eukaryotic proteins
NetPhosBac: Generic phosphorylation sites in bacterial proteins
ProP: Arginine and lysine propeptide cleavage sites in eukaryotic protein sequences
2.3 Secondary structure prediction:
APSSP2: Advanced Protein Secondary Structure Prediction Server
2.4 Turn prediction:
BhairPred: SVM based method for prediction of beta-hairpins in proteins
BTEVAL: Evaluation of beta turns prediction methods
BetaTPred: predicting ß-turns in a protein from the amino acid sequence
BetaTPred2: Prediction
of ß-turns in proteins using neural
networks and multiple alignments
Betaturns: Prediction of beta-turn types
AlphaPred: predicts the alpha turn residues in the given protein sequence
GammaPred: predicts the gamma turn residues in the given protein sequence
RELEVANCE:
Useful application of DNA-binding residues prediction would be the identification of proteins that bind to DNA. Recognition of probable binding sites both on the protein and the DNA will go a long way in diagnosing the basis of these interactions. Their discovery can help lead subsequent works such as site-directed mutagenesis and constrained macromolecular docking. Prediction of functional sites to act as filters in a predictive scheme for docking can be as effective as manually introducing biological constraints.
The identification of DNA-binding sites can also assist in prediction of DNA-binding behaviour of a protein. This is similar in spirit to other studies that assign functions to a protein on the basis of functional sites discovered on its surface, such as protein-protein and protein-DNA interaction sites.
3. Prediction at peptide/epitope level:
The potential importance of epitope identification in developing vaccines against infectious, immune and other antigen-related diseases, epitopes are studied widely by researchers in various fields, and a large expansion of databases, predictive methods and software focussing on different types of epitopes has been witnessed. The average immunologists are overwhelmed with such a broad array of immunological analysis tools that are highly specific in use, not well understood or defined, tested on limited data and not publicly accessible
Epitope prediction dates back to 1981 when the first B cell epitope prediction method was developed by Hopp and Woods. Since then many more methods have been developed or adapted from other computational tools; for example B cell epitope prediction and T cell epitope prediction. Despite the early start, however, prediction systems for B cell epitopes are still in their infancy.
General epitope prediction methods
- Sequence-based epitope prediction
Sequence-based method utilises the notion that sequence dictates structure and identical structure in turn leads to identical functions. T cell epitopes have a common sequence pattern or motif, as well as MHC allele specificity determining subpatterns.
To make useful, informative epitope prediction, epitope physicochemical properties are also used, such as exposed surface, accessibility, flexibility, hydrophilicity, charge, number of proline residues, the proximity of the segment towards the C- or N-terminal of the protein, etc. Due to the enormous number of physicochemical properties that are associated with epitopes, simpler quantitative descriptors of amino acid properties are sometimes used to simplify computation.
Techniques, such as binding motifs, quantitative matrices (QM), virtual matrices, machine learning algorithms (ANN, HMM, SVM), evolutionary algorithms, linear programming, etc. are used to identify the binding peptide. They all have their relative advantages and disadvantages. For example, in a comparative study, Yu et al. suggested that motifs give the most accurate MHC-peptide binding predictions with a limited dataset, but as the data volume increases, machine learning predictions become more reliable.
- Structure-based epitope prediction
The structure-based prediction model bases on 3D protein structure to screen potential binders. Structural similarity between query protein and template proteins are used to predict epitopes of interest.
- Hybrid prediction methods: combining sequential with structural analysis
Given the poor performance of epitope predictors based on sequence or structure analysis, it is clear that any single method cannot accurately predict epitopes. Consequently, some researchers turned to building predictive methods taking advantage of both sequential and structural information. For example, a new method which integrates 3D protein structure with physicochemical properties of amino acids using machine learning methods like Hidden Markov Model (HMM), supporting vector machine (SVM), ANN, etc. improved the prediction precision to a though small but significant degree. Like structural-based approach, its further development is hampered by the limited availability of 3D structure data of antigens and true negative datasets, both to construct better predictors and evaluate the algorithms. There is also the possibility of false positives because different antibodies have overlapping binding sites.
METHODS
1. ProPred: predicting binders of 51 HLA-DR (MHC class II of human) alleles.
2. Propred1: binding peptides of MHC class I alleles. Matrix based methods
3. nHLAPred: Promiscuous MHC class I restricted T cell epitopes: ANN, QM
4. CTLPred: predicting cytotoxic T lymphocyte (CTL) epitopes in an antigenic sequence: SVM, QM, ANN
5. TAPPred: predicting TAP binding peptide in a protein
6. BcePred: Prediction of linear B-cell epitopes, using physico-chemical properties
7. ABCPred: predict B cell epitope(s) in an antigen sequence, using artificial neural network
8. Pcleavage: SVM based method for Proteosome cleavage prediction
9. MMBpred: predict mutated high affinity and promiscuous MHC class-I binding peptides from protein sequence
10. HLA-DR4Pred: an SVM and ANN based HLA-DRB1*0401(MHC class II alleles) binding peptides prediction method
RELEVANCE:
The implication of epitope prediction in both public health and basic scientific research is vast. It is applicable to all epitope-related research, such as discovery of peptide candidate for subunit vaccines, autoimmune diseases study, allergy treatment, protein structural study, experiment design, etc. Developing epitope predictive methods and software to identify and map potential epitopes from an antigen protein is vital to contest the immune and infectious diseases. Drug development is the major financial drive for epitope prediction. Epitope-based vaccines have been shown to have promising results and confer protection to animal models in clinical trials, supporting the prophylactic, therapeutic and protective effects of these vaccines. The advantages of subunit vaccines over other types of vaccines are pronounced. Therefore, huge resources are being channelled into developing subunit vaccines against important, intractable diseases such as cancer, HIV/AIDS, HCV and many other infectious, viral and immune diseases.
However, despite its huge implications in public health, security and scientific arena, epitope prediction tools may be abused by terrorists to make biochemical weapons, and accelerate pathogen evolution and mutation. Another concern is that the application of epitope predictive software in discovering epitopes bias subsequent predictors, as researchers would normally narrow peptide targets by predicting possible epitopes first and then conduct experiments to discover epitopes, which in turn will be analysed to develop other epitope predictive software.
4. Prediction based on signal sequences:
Protein localization is important as protein function may be localized to specific areas inside the cell or within cellular organelles. These bioinformatics programs and databases contain information and are able to predict where a protein may be localized based on signal sequences or localization sequences contained within the protein. Methods involving the recognition of N-terminal signal sequences; as the strong biological implication because the signal sequence specifying the cellular location of a protein is located at the N-terminus (Emanuelsson et al., 2000 and Reczko and Hatzigerrorgiou, 2004). However, it is difficult to recognize underlying features from a highly diverged signal sequence and to vectorize those features.
METHODS:
pTARGET: (Guda and Subramaniam, 2005) uses amino acid composition and localization-specific Pfam domains to assign a eukaryotic protein to one of nine localization sites.
SecretomeP: (Bendtsen et al, 2004) predicts eukaryotic proteins which are secreted via a non-traditional secretory mechanism.
SignalP: (Bendtsen et al, 2004) predicts traditional N-terminal signal peptides in both prokaryotic and eukaryotic proteins.
TargetP: (Emanuelsson et al, 2000) predicts the presence of signal peptides, chloroplast transit peptides, and mitochondrial targeting peptides for plant proteins, and the presence of signal peptides and mitochondrial targeting peptides for eukaryotic proteins.
ChloroP: Chloroplast transit peptides and their cleavage sites in plant proteins
5. Prediction based on Motifs:
The rapid increase in genomic information requires new techniques to infer protein function and predict protein-protein interactions. Bioinformatics identifies modular signalling domains within protein sequences with a high degree of accuracy. In contrast, little success has been achieved in predicting short linear sequence motifs within proteins targeted by these domains to form complex signalling networks. Predictions from database searches for proteins containing motifs matching two different domains in a common signaling pathway provide a much higher success rate. This technology facilitates prediction of cell signalling networks within proteomes, and could aid in the identification of drug targets for the treatment of human diseases.
Techniques used for finding motifs in given protein sequences:
MEME: tool for discovering motifs in a group of related DNA or protein sequences.
Prosite: This program allows to scan a protein sequence (either from Swiss-Prot or TrEMBL or provided by the user) for the occurrence of patterns and profiles stored in the PROSITE database, or to search protein databases with a user-entered pattern.
PRINTS: is a compendium of protein fingerprints. A fingerprint is a group of conserved motifs used to characterise a protein family; its diagnostic power is refined by iterative scanning of a SWISS-PROT/TrEMBL composite. Usually the motifs do not overlap, but are separated along a sequence, though they may be contiguous in 3D-space.
METHODS:
Pseapred: prediction of secretory proteins of P.falciparum method employs MAST technique along with PSI-BLAST and PSSM.
TBpred: prediction server that predicts four subcellular localization (cytoplasmic, integral membrane, secretory and membrane attached by lipid anchor) of mycobacterial proteins .It is SVM based method that exploits different features of protein such as amino acid composition, dipeptide composition and position specific scoring matrix (PSSM). Along with SVM other techniques like profile HMM and MEME/MAST motif based studies were also applied. Moreover a hybrid approach combining the PSSM based SVM model and the MEME/MAST model has been incorporated.
6. Prediction based on Domains:
Protein domain prediction is important for protein structure prediction, structure determination, function annotation, mutagenesis analysis and protein engineering.
Protein domains are structural, functional and evolutionary units of proteins. The prediction of domains from sequence information can improve tertiary structure prediction; enhance protein function annotation, aid structure determination and guide protein engineering and mutagenesis.
The identification of domains within a protein sequence is an important precursor for a range of methods. Protein structural determination method such as X-ray crystallography and NMR has size limitations which limit their use - they are often employed more successfully when solving smaller domain units rather than whole chains.
METHODS:
MITPred: method for predicting the proteins which are destined to localize in mitochondria. In this method Domain search technique is also employed using my HMMER (hidden Markov Models based search) along with BLAST and SVM.
RELEVANCE:
Domains provide one of the most valuable information for the prediction of protein structure, function, evolution and design. Accurate prediction of domain boundaries forms a basis of many types of protein research. New proteins such as chimeric proteins can be created as they are composed of multifunctional domains (Suyama & Ohara, 2003). The search method for templates used in comparative modeling can also be optimized by the delineation of domain boundaries (Contreras-Moreira & Bates, 2002). As for threading methods, the domain boundary prediction can improve its performance by enhancing the signal-to-noise ratio (Wheelan et al., 2000). Accurate identification of domain boundaries for homologous domains plays a key role for reliable multiple sequence alignment (Gracy & Argos, 1998).
7. Prediction based on Profiles:
Classic profile-based prediction worked well for early single-issue, in-order execution processors, but fails to accurately predict the performance of modern processors. The major reason is that modern processors can issue and execute several instructions at the same time, sometimes out of the original order and cross the boundary of basic blocks.
Prosite is a method of determining what is the function of uncharacterized proteins translated from genomic or cDNA sequences. It consists of a database of biologically significant sites and patterns formulated in such a way that with appropriate computational tools it can rapidly and reliably identify to which known family of protein (if any) the new sequence belongs.
A profile, or weight matrix, is a table of position-specific amino acid weights and gap costs. These numbers (also referred to as scores) are used to calculate a similarity score for any alignment between a profile and a sequence, or parts of a profile and a sequence. An alignment with a similarity score higher than or equal to a given cut-off value constitutes a motif occurrence. As with patterns, there may be several matches to a profile in one sequence, but multiple occurrences in the same sequences must be disjoint (non-overlapping) according to a specific definition included in the profile.
METHODS:
TBpred: prediction server that predicts four subcellular localization (cytoplasmic, integral membrane, secretory and membrane attached by lipid anchor) of mycobacterial proteins. It is SVM based method that exploits different features of protein such as amino acid composition, dipeptide composition and position specific scoring matrix (PSSM). Along with SVM other techniques like profile HMM and MEME/MAST motif based studies were also applied. Moreover a hybrid approach combining the PSSM based SVM model and the MEME/MAST model has been incorporated.
PPrint: is a web-server for predicting RNA-binding residues of a protein. The prediction is done by SVM model trained on PSSM profile generated by PSI-BLAST search of 'nr' protein database.
PFMpred: Predicting mitochondrial proteins of P.falciparum
ESLPred2: Prediction of subcellular localization of eukaryotic proteins