Protein General Modules
In this chapter we have described the small programs developed at our group; these programs can be used as building block to develop complex prediction modules. The question arises how it is different then existing software libraries or modules like BioPERL. InBioPER or similar packages one need to have knowledge of computer programming in order to uses these modules/subroutines. In GPSR package we have developed small programs, which can be run by any person have little knowledge of computers. Following are important programs included in this package.
|
Program |
Purpose |
|
· fasta2sfasta |
Convert fasta format to single fasta format |
|
· pro2aac |
To calculate amino acid composition of protein |
|
· pro2aac_nt |
To calculate amino acid composition of N-terminal (nt) residues of a protein |
|
· pro2aac_ct |
To calculate amino acid compositionof C-terminal (ct) residues of a protein |
|
· pro2aac_rest.pl |
To calculate amino acid composition of a protein after removing N-, and C-terminal residues |
|
· pro2aac_split |
To calculate split amino acid composition (SSAC) of a protein |
|
· pro2dpc |
To calculate dipeptide composition of protein |
|
· pro2dpc_nt |
To calculate dipeptide composition of N-terminal (nt) residues of a protein |
|
· pro2dpc_ct |
To calculate dipeptide composition of C-terminal (ct)residues of a protein |
|
· pro2tpc |
To calculate tripeptide composition of protein |
|
· add_cols |
To add columns of two files |
|
· col2svm |
To generating SVM_light input format |
|
· col_mult |
To multiplying each column of input file with a number |
|
· col_mult_sel |
To multiplying selective columns with a number |
|
· perl col_rem |
To remove selective columns from a file |
|
· col_ext |
To extract selective columns from a file |
|
· col_corr |
To compute correlation co-efficient between two column |
|
· col_avg |
To calculate average column of two files |
|
· seq2pssm_imp |
To calculate PSSM matrix in column format without any normalization |
|
· pssm_n1 |
To normalize pssm profile based on 1/(1+e-x) formula |
|
· pssm_n2 |
To normalize pssm profile based on (numb -min)/(max -min) formula |
|
· pssm_n3 |
To normalize pssm profile based on (numb -min)*100/(max -min) formula |
|
· pssm_n4 |
To normalize pssm profile based on 1/(1+e-(x/100) formula |
|
· pssm_comp |
To compute PSSM composition (400 points) |
|
· col_sig |
Significance of columns in two column files |
|
· pssm2pat |
To generate patterns of given size from PSSM matrix |
|
· pssm_smooth |
To designed smooth pssm profile for plot |
|
· seq2motif |
To create motifs by sliding window of user defined length with option of adding terminal X |
|
· motif2bin |
To make binary input from the multifasta motif file |
|
· blast_similarity |
To perform blast |
Title |
Description |
Fasta format |
fasta2sfasta (Convert fasta format to single fasta format)(Pearon format) is used to represent peptide sequences or nucleic acid sequences using single-letter codes. It begins with a single-line description, followed by lines of sequence data. The description line is distinguished from the sequence data by a greater-than (">") symbol. |
Single fasta format |
Our programs use input sequence in single fasta format. Therefore, fasta file should first convert into single fasta format. In the single fasta format the description and sequence data merged into single line. Two hash marks (##) were present to distinguish description and sequence data. |
Usage |
fasta2sfasta –i seq.fa -o seq.sfa |
-i |
Input file name having sequence in fasta format |
-o |
Output file name that gives sequence in single fasta format |
seq.fa |
>seq_1MRNRGFGRRELLVAMAMLVSVTGCARHASGARPASTTLPAGADLADRFAELERRYDARLGVYVPATGTTAAIE>seq_2ACGRGFGVKLACNMNNACRTYFSDVAMAMLVSVTGCARHASGARPASTTLPAGADLADIEYRADERFAFCSTF |
seq.sfa |
>seq_1##MRNRGFGRRELLVAMAMLVSVTGCARHASGARPASTTLPAGADLADRFAELERRYDARLGVYVPATGTTAAIE>seq_2##ACGRGFGVKLACNMNNACRTYFSDVAMAMLVSVTGCARHASGARPASTTLPAGADLADIEYRADERFAFCSTF |
Title |
Description |
|||
|
|
pro2aac (To calculate amino acid composition of protein)The amino acid composition in a protein is simply the percentage of the different amino acids represented in a particular protein. The aim of calculating the composition of proteins is to transform the variable length of protein sequences to fixed length feature vectors. This is an important and most crucial step during classification of proteins using machine-learning techniques because they require fixed length patterns. In addition the conversion of a protein sequence to a vector of 20 dimensions using amino acid composition will encapsulate the properties of the protein into the vector.
The composition of all 20 natural amino acids were calculated by using the following equation
Where i can be any amino acid |
|||
|
Usage |
pro2aac -i seq.sfa -o seq.out |
|||
|
-i |
Input file name contains single fasta format |
|||
|
-o |
Output file name gives amino acid composition |
|||
|
seq.sfa |
>seq_1##MRNRGFGRRELLVAMAMLVSVTGCARHASGARPASTTLPAGADLADRFAELERRYDARLGVYVPATGTTAAIE >seq_2##ACGRGFGVKLACNMNNACRTYFSDVAMAMLVSVTGCARHASGARPASTTLPAGADLADIEYRADERFAFCSTF |
|||
|
seq.out |
# Amino Acid Composition of proteins # A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y, 19.18, 1.37, 4.11, 5.48, 2.74, 9.59, 1.37, 1.37, 0.00, 9.59, 4.11, 1.37, ...... 2.74, 19.18, 6.85, 5.48, 2.74, 6.85, 8.22, 1.37, 1.37, 1.37, 5.48, 4.11, 4.11, ...... 2.74,
|
|||
|
Vector |
20 dimension (i.e 20 types of amino acid composition is generated) |
Title |
Description |
|||
|
|
pro2aac_nt (To calculate amino acid composition of N-terminal (nt) residues of a protein)It is well known that some proteins having N-terminal signal sequence which is responsible to transport whole protein into their specific subcellular compartment like, lysosome, endoplasmic reticulum, mitochondria, and chloroplast. Evidences indicate that divergent N-terminal sequences also do influence catalytic behavior, protein-protein interactions, and intracellular distributions of enzymes. Report shows that N-terminal signal sequence can vary from 13 to 36 amino acid residues in length and having all the information needed to localize into specific location. Therefore, N-terminal information could be exploited by using amino acid composition feature to predict subcellular protein. For example:N 5 nt C |
|||
Usage |
pro2aac_nt -i seq.sfa -o seq.out -n 5 |
|||
-i |
Input file name |
|||
-o |
Output file name |
|||
-n |
Number of residues to calculate composition from N-terminal |
|||
seq.sfa |
>seq_1##MRNRGFGRRELLVAMAMLVSVTGCARHASGARPASTTLPAGADLADRFAELERRYDARLGVYVPATGTTAAIE>seq_2##ACGRGFGVKLACNMNNACRTYFSDVAMAMLVSVTGCARHASGARPASTTLPAGADLADIEYRADERFAFCSTF |
|||
Seq.out |
# Amino Acid Composition of 5 n-terminal residues of proteins# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y,0.00, 0.00, 0.00, 0.00, 0.00, 20.00, 0.00, 0.00, 0.00, 0.00, 20.00 ..... 0.00,20.00, 20.00, 0.00, 0.00, 0.00, 40.00, 0.00, 0.00, 0.00, 0.00, ..... 0.00, |
|||
Vector |
20 dimension |
Title |
Description |
|||
|
|
pro2aac_ct (To calculate amino acid composition of C-terminal (ct) residues of a protein)While the N-terminus of a protein often contains targeting signals, the C-terminus can contain retention signals for protein sorting. The most common ER retention signal is the amino acid sequence -KDEL (or -HDEL) at the C-terminus, which keeps the protein in the endoplasmic reticulum and prevents it from entering the secretory pathway. The C-terminus of proteins can be modified post-translationally, most commonly by the addition of a lipid anchor to the C-terminus that allows the protein to be inserted into a membrane without having a transmembrane domain. The c-terminal domain of RNA polymerase II typically consists of up to 52 repeats of the sequence Tyr-Ser-Pro-Thr-Ser-Pro-Ser. Other proteins often bind the C-terminal domain of RNA polymerase in order to activate polymerase activity. It is the protein domain, which is involved in the initiation of DNA transcription, the capping of the RNA transcript, and attachment to the spliceosome for RNA splicing. Therefore the information at C-terminal in could be utilized using amino acid composition feature to predict different classes of proteins. For example:N 5nt C |
|||
Usage |
pro2aac_ct -i seq.sfa -o seq.out -n 5 |
|||
-i |
Input file name |
|||
-o |
Output file name |
|||
-n |
Number of residues to calculate composition from C-terminal |
|||
seq.sfa |
>seq_1##MRNRGFGRRELLVAMAMLVSVTGCARHASGARPASTTLPAGADLADRFAELERRYDARLGVYVPATGTTAAIE>seq_2##ACGRGFGVKLACNMNNACRTYFSDVAMAMLVSVTGCARHASGARPASTTLPAGADLADIEYRADERFAFCSTF |
|||
seq.out |
# Amino Acid Composition of 5 c-terminal residues of proteins# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y,40.00, 0.00, 0.00,20.00, 0.00, 0.00, 0.00,20.00, 0.00, 0.00, 0.00, 0.00, 0.00, .... 0.00,0.00,20.00, 0.00, 0.00,40.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, ...... 0.00, |
|||
Vector |
20 dimension |
Title |
Description |
|||
|
|
pro2aac_rest (To calculate amino acid composition of a protein after removing N-, and C-terminal residues)This program is used to calculate the composition of remaining part of a protein after removing specific residues from N-, and C-terminus. Transmembrane proteins having membrane spanning signal in the middle of protein. This program can be used to calculate the amino acid composition of middle part and successfully used in classification family of proteins. For example:N 5 nt 5 nt C |
|||
Usage |
pro2aac_rest -i seq.sfa -o seq.out -n 5 –c 5 |
|||
-i |
Input file name |
|||
-o |
Output file name |
|||
-n |
Number of residues removed from N-terminal |
|||
-c |
Number of residues removed from C-terminal |
|||
seq.sfa |
>seq_1##AAAAACCCCCGGGGG>seq_2##CCCGCAAAAASNMKL |
|||
seq.out |
# Amino Acid Composition of protein after removing 5 n-terminal and 5 c-terminal residues# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y,0.00, 100.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, ..... 0.00,100.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00 ..... 0.00, |
|||
Vector |
20 dimension |
Title |
Description |
|||
|
|
pro2aac_split (To calculate split amino acid composition (SSAC) of a protein)It has been reported that some sequence motifs are present into specific region of a protein. Therefore, instead of computing the composition of whole sequence it is useful to split the sequence into different equal parts. Composition of each part is separately calculated, thus feature of region specific motifs is utilized, and added to each other. Some reports show that is increases the prediction accuracy after using this strategy. The advantage of SSAC over standard amino acid composition is that it provides greater weight to proteins that have a signal at either the N or C terminus. For Example:N C |
|||
Usage |
pro2aac_split -i seq.sfa -o seq.out -n 3 |
|||
-i |
Input file name |
|||
-o |
Output file name |
|||
-n |
Number of parts split into, here 3 i.e. three equal parts of whole protein |
|||
seq.sfa |
>seq_1##AAAAACCCCCGGGGG>seq_2##CCCGCAAAAASNMKL |
|||
seq.out |
# Amino Acid Composition of 3 equal parts of proteins# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y,5.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 5.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 5.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00,0.00, 4.00, 0.00, 0.00, 0.00, 1.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 5.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 0.00, 1.00, 1.00, 1.00, 1.00, 0.00, 0.00, 0.00, 1.00, 0.00, 0.00, 0.00, 0.00, |
|||
Vector |
60 dimension (20*3 parts) |
Title |
Description |
||||||
|
|
pro2dpc (To calculate dipeptide composition of protein)The dipeptide composition in a protein is simply the percentage of the different adjacent pairs of amino acids represented in a particular protein. The aim of calculating the composition of proteins is to transform the variable length of protein sequences to fixed length feature vectors. This is an important and most crucial step during classification of proteins using machine-learning techniques because they require fixed length patterns. In addition the conversion of a protein sequence to a vector of 400 dimensions using dipeptide composition will encapsulate the properties of the neighboring amino acids.
The composition of all 400 natural amino acids were calculated by using the following equation
Where dpep (i) is fraction or composition of dipeptide type i. Di and N are the number of dipeptide of type i and number of residues in protein i, respectively. |
||||||
Usage |
pro2dpc -i seq.sfa -o seq.out |
||||||
-i |
Input file name |
||||||
-o |
Output file name |
||||||
seq.sfa |
>seq_2##AAAAACCCCCGGGGG |
||||||
seq.out |
#AA , AC , AD ,….., CC ,….., CG , ….. , GG ,….., YY,28.571, 7.143, 0.000, ….., 28.571,….., 7.143, ….., 28.571,….., 0.000 |
||||||
Vector |
400 dimension (20*20) |

Title |
Description |
|
|
pro2dpc_nt (To calculate dipeptide composition of N-terminal (nt) residues of a protein)It is well known that some proteins having N-terminal signal sequence which is responsible to transport whole protein into their specific subcellular compartment like, lysosome, endoplasmic reticulum, mitochondria, and chloroplast. Evidences indicate that divergent N-terminal sequences also do influence catalytic behavior, protein-protein interactions, and intracellular distributions of enzymes. Report shows that N-terminal signal sequence can vary from 13 to 36 amino acid residues in length and having all the information needed to localize into specific location. Therefore, N-terminal information could be exploited by using dipeptide composition feature to predict subcellular protein. |
Usage |
pro2dpc_nt -i seq.sfa -o seq.out -n 5 |
-i |
Input file name |
-o |
Output file name |
-n |
Number of residues to calculate dipeptide composition from N-terminal |
seq.sfa |
>seq_2##AAAAACCCCCGGGGG |
Seq.out |
# Dipeptide composition of 5 n-terminal residues of proteins#AA , AC , AD ,….., CC ,….., CG , ….. , GG ,….., YY,100.00, 0.000, 0.000, ….., 00.000,….., 0.000, ….., 00.000,….., 0.000 |
Vector |
400 dimension |
Title |
Description |
|
|
pro2dpc_ct (To calculate dipeptide composition of C-terminal (ct) residues of a protein)While the N-terminus of a protein often contains targeting signals, the C-terminus can contain retention signals for protein sorting. The most common ER retention signal is the amino acid sequence -KDEL (or -HDEL) at the C-terminus, which keeps the protein in the endoplasmic reticulum and prevents it from entering the secretory pathway. The C-terminus of proteins can be modified post-translationally, most commonly by the addition of a lipid anchor to the C-terminus that allows the protein to be inserted into a membrane without having a transmembrane domain. The c-terminal domain of RNA polymerase II typically consists of up to 52 repeats of the sequence Tyr-Ser-Pro-Thr-Ser-Pro-Ser. Other proteins often bind the C-terminal domain of RNA polymerase in order to activate polymerase activity. It is the protein domain, which is involved in the initiation of DNA transcription, the capping of the RNA transcript, and attachment to the spliceosome for RNA splicing. Therefore the information at C-terminal in could be utilized using dipeptide composition feature to predict different classes of proteins. |
Usage |
pro2dpc_ct -i seq.sfa -o seq.out -n 5 |
-i |
Input file name |
-o |
Output file name |
-n |
Number of residues to calculate dipeptide composition from C-terminal |
seq.sfa |
>seq_2##AAAAACCCCCGGGGG |
Seq.out |
# Dipeptide composition of 5 n-terminal residues of proteins#AA , AC , AD ,….., CC ,….., CG , ….. , GG ,….., YY,100.00, 0.000, 0.000, ….., 00.000,….., 0.000, ….., 100.000,….., 0.000 |
Vector |
400 dimension |
Title |
Description |
||||||
|
|
pro2tpc (To calculate tripeptide composition of protein)The tripeptide composition in a protein is simply the percentage of the three adjacent amino acids represented in a particular protein. The aim of calculating the composition of proteins is to transform the variable length of protein sequences to fixed length feature vectors. This is an important and most crucial step during classification of proteins using machine-learning techniques because they require fixed length patterns. In addition the conversion of a protein sequence to a vector of 8000 dimensions using tripeptide composition will encapsulate the properties of the neighboring amino acids.
The composition of all 8000 natural amino acids were calculated by using the following equation
|
||||||
Usage |
pro2tpc -i seq.sfa -o seq.out |
||||||
-i |
Input file name |
||||||
-o |
Output file name |
||||||
seq.sfa |
>seq_2##AAAAACCCCCGGGGG |
||||||
Seq.out |
# Tripeptide Composition of Protein#AAA ,AAC ,AAD ,AAE ,AAF , …..,,YYW ,YYY23.0769 , 7.6923, 0.000, 00.000, 0.000, ….., 0.000 , 0.000 |
||||||
Vector |
8000 dimension |
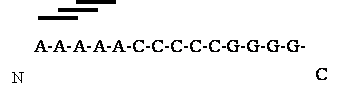
Title |
Description |
|
|
col_add (To add columns of two files)It is used to make a hybrid method. In this two different features (e.g. amino acid composition, and dipeptided) of a sequence are added to make a more informative hybrid features. |
Usage |
add_cols -i se1.out -c se2.out -o seq.out |
-i |
Input file (first column file for add) |
-c |
Input file (second column file for add) |
-o |
Output file name |
se1.out |
Amino Acid Composition of proteins# A , C , D , E , F , G , ….., Y,33.33,33.33, 0.00, 0.00, 0.00,33.33, ….., 0.00 |
se2.out |
# Dipeptide Composition of Protein#AA , AC , ….., YY28.571,7.143…..,0.00 |
seq.out |
# Amino Acid Composition of proteins # Dinucleic Composition of Protein# A , C , D , E , F , G , …., #AA , AC, .…, YY33.33,33.33, 0.00, 0.00, 0.00, 33.33,……, 28.571,7.143…..,0.00 |
Vector |
420 (20 for amino acid + 400 dipeptide composition) |
Title |
Description |
|
|
col_mult (To multiplying each column of input file with a number)This program is used to multiply each column of input file with a specific number. This is used especially in the hybrid case to make the features equal weight. Suppose one wants to make a hybrid file of amino acid and dipeptide composition. If amino acid and dipeptide composition was added directly the values of mononucleotide is very high with respect to dinucleotide. Thus performance of SVM will be nearly similar to the performance of amino acid because the weight of dipeptide is diluted. But when we multiply the amino acid with 10 or dipeptide with 0.1 and then added to each other. There is chance that performance will increase. |
Usage |
col_mult -i se1.out -o se1_mult -n 0.1 |
-i |
Input file name |
-o |
Output file name |
-n |
Number with which column is multiplying |
se1.out |
Amino Acid Composition of proteins# A , C , D , E , F , G , ….., Y,33.33, 33.33, 0.00, 0.00, 0.00, 33.33, ….., 0.00 |
se1_mult |
3.333000, 3.333000, 0.000000, 0.000000, 0.000000, 3.333000,….. , 0.000000, |
Vector |
Same as in input file |
Title |
Description |
|
|
col2svm (To generating SVM_light input format)This program can convert composition output file into a format used in SVM training. In SVM format, (1) starts with +1 or –1 denotes class of sequence positive or negative respectively. (2) A numerical order is given before each value. |
Usage |
col2svm -i se1.out -o svm.out -s +1 |
-i |
Input file name |
-o |
Output file name |
-s |
Class for svm (+1 or –1) |
se1.out |
Amino Acid Composition of proteins# A, C, D, E, F, G,… Y,33.33, 33.33, 0.00, 0.00, 0.00, 33.33, ….., 0.00 |
svm.out |
+1 1:33.330000 2:33.330000 3:0.000000 4:0.000000 5:0.000000 6:33.330000 ……. 20:0.000000 |
Vector |
20 dimension |
Title |
Description |
|
|
col_mult_sel (To multiplying selective columns with a number)Instead of multiplying whole column, here only column from 1 to 3 are multiplied with specific number (10). |
Usage |
col_mult_sel -i se1.out -o se1_mult -n 10 -a 1 -b 3 |
-i |
Input file name |
-o |
Output file name |
-n |
Number with which column is multiplying |
-a |
Number of starting column (eg 1) |
-b |
Number of last column (eg 3) |
se1.out |
Amino Acid Composition of proteins# A , C , D , E , F , G , ….., Y,33.33, 33.33, 0.00, 0.00, 0.00, 33.33, ….., 0.00 |
se1_mult |
333.300000, 333.300000, 0.000000, 0.000000, 0.000000, 33.330000,….., 0.000000 |
Vector |
Same as in input file |
Title |
Description |
|
|
col_rem (To remove selective columns from a file)This program is used to remove specific column from files. You can remove the composition of A and C from whole file to check the importance of these amino acids in prediction methods. |
Usage |
perl col_rem -i seq.out -o seq.rm -a 1 -b 2 |
-i |
Input file name |
-o |
Output file name |
-a |
Number of starting column (eg 1) to removed |
-b |
Number of last column (eg 3) removed |
seq.out |
# Amino Acid Composition of proteins# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y,18.60, 2.33, 4.65, 5.81, 5.81, 8.14, 1.16, 1.16, 0.00, 8.14, 3.49, 1.16, 3.49, 0.00, 13.95, 4.65, 8.14, 5.81, 0.00, 3.49, |
Seq_rm |
5.810000,5.810000,8.140000,1.160000,1.160000,0.000000,8.140000,3.490000, 1.160000,3.490000,0.000000,13.950000,4.650000,8.140000,5.810000,0.000000,3.490000 |
Vector |
Total number = Total columns in the input file – total number of removed columnE.g.(17=20-3) |
Title |
Description |
|
|
col_ext (To extract selective columns from a file)This program only takes specific column from a file. In this example we only take the feature of amino acid composition of F, G, H, I, and K as an input for SVM. |
Usage |
col_ext -i seq.out -o seq.ext -a 5 -b 10 |
-i |
Input file name |
-o |
Output file name |
-a |
Number of starting column (eg 5) to take |
-b |
Number of last column (eg 10) to take |
seq.out |
# Amino Acid Composition of proteins# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y,18.60, 2.33, 4.65, 5.81, 5.81, 8.14, 1.16, 1.16, 0.00, 8.14, 3.49, 1.16, 3.49, 0.00, 13.95, 4.65, 8.14, 5.81, 0.00, 3.49, |
Seq.ext |
5.81, 8.14, 1.16, 1.16, 0.00, 8.14 |
Vector |
Total number = Total number of column selected from input fileEg(6=from 5 to 10 colum) |
Title |
Description |
|
|
col_corr (To compute correlation co-efficient between two column)Correlation co-efficient indicates the strength and direction of a linear relationship between two random variables. The correlation varies between –1 to 1. The closer the coefficient is to either -1 or 1, the stronger the correlation between the variables. Value of 1 in the case of an increasing linear relationship, -1 in the case of a decreasing linear relationship, 0 in case no correlation. Example shows the correlation between amino acid A and G in the file. |
Usage |
col_corr -i pos -a 1 -b 6 |
-i |
Input file name |
-a |
Number of column (eg 1) |
-b |
Number of column (eg 6) |
pos |
# Amino Acid Composition of proteins# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y,15.31, 1.30, 7.49, 3.91, 2.28, 9.45, 1.63, 3.26, 1.95, 10.10, 1.95, 2.28, 5.54, 2.28, 8.14, 3.91, 8.47, 6.84, 0.98, 2.93,15.31, 1.30, 7.49, 3.91, 2.28, 9.45, 1.63, 3.26, 1.95, 10.10, 1.95, 2.28, 5.54, 2.28, 8.14, 3.91, 8.47, 6.84, 0.98, 2.93,12.83, 1.60, 8.56, 2.14, 2.67, 6.95, 6.95, 2.67, 1.60, 9.09, 3.74, 3.74, 6.42, 6.42, 4.28, 5.35, 6.42, 5.35, 1.07, 2.14,12.30, 1.60, 8.56, 2.14, 2.67, 6.95, 6.95, 2.67, 1.60, 9.09, 3.74, 3.74, 6.42, 6.42, 4.28, 5.35, 6.42, 5.88, 1.07, 2.14,13.76, 0.53, 4.76, 4.23, 3.70, 5.29, 1.06, 3.17, 2.12, 8.47, 3.17, 3.70, 13.76, 1.59, 4.76, 9.52, 8.99, 5.82, 1.06, 0.53, |
output |
0.749 (a positive correlation between column 1 and 6) |
Vector |
Total number = Total number of column selected from input fileE.g.(6=from 5 to 10 colum) |
Title |
Description |
|
|
col_avg (To calculate average column of two files)In this case composition value of each column of a file is added to its corresponding column of another file and means value is calculated. It can be used to generate an average feature of two different files (belonging from same family of protein) to make input in machine learning techniques. For instance, 15.31 (1st column of file pos1) + 6.87 (1st column of file pos2) = 22.18/2 = 11.09 (file out).Note: Each file should have equal number of columns and rows |
Usage |
col_avg -a pos1 -b pos2 -o out |
-a |
First input file name |
-b |
Second input file name |
-o |
Output file name |
pos1 |
# Amino Acid Composition of proteins# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y,15.31, 1.30, 7.49, 3.91, 2.28, 9.45, 1.63, 3.26, 1.95,10.10, 1.95, 2.28, 5.54, 2.28, \8.14, 3.91, 8.47, 6.84, 0.98, 2.93,15.31, 1.30, 7.49, 3.91, 2.28, 9.45, 1.63, 3.26, 1.95,10.10, 1.95, 2.28, 5.54, 2.28, \8.14, 3.91, 8.47, 6.84, 0.98, 2.93,12.83, 1.60, 8.56, 2.14, 2.67, 6.95, 6.95, 2.67, 1.60, 9.09, 3.74, 3.74, 6.42, 6.42, \4.28, 5.35, 6.42, 5.35, 1.07, 2.14, |
pos2 |
# Amino Acid Composition of proteins# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y,6.87, 1.29, 6.87, 2.15, 0.86, 6.87, 1.72, 4.72, 5.58, 9.87, 1.29, 5.15, 4.72, 5.15, 1.72,13.73, 9.01,10.30, 1.72, 0.43,9.87, 1.29, 7.30, 3.00, 0.86, 8.58, 1.29, 4.72, 4.29, 9.87, 0.86, 3.43, 5.15, 4.29, 3.43,11.16, 7.73, 10.73, 1.72, 0.43,12.64, 1.10, 4.40, 1.10, 2.75, 9.34, 0.55, 6.59, 3.30, 9.34, 2.20, 6.59, 6.04, 5.49, 4.40, 6.59, 7.14, 9.34, 0.55, 0.55, |
out |
11.09; 1.295; 7.18; 3.03; 1.57; 8.16; 1.675; 3.99; 3.765; 9.985; 1.62; 3.715; 5.13; 3.715; 4.93; 8.82; 8.74; 8.57; 1.35; 1.68; 012.59; 1.295; 7.395; 3.455; 1.57; 9.015; 1.46; 3.99; 3.12; 9.985; 1.405; 2.855; 5.345; 3.285; 5.785; 7.535; 8.1; 8.785; 1.35; 1.68; 012.735; 1.35; 6.48; 1.62; 2.71; 8.145; 3.75; 4.63; 2.45; 9.215; 2.97; 5.165; 6.23; 5.955; 4.34; 5.97; 6.78; 7.345; 0.81; 1.345; 0 |
Vector |
Total number of column (same as in input file) |
Title |
Description |
|
|
col_sig (significance of columns in two column files)This program used to calculate significant of each column in two different file. If any one want to compare the positive and negative file of amino acid composition. Like Differences in positive-negative, its significance, average of each colomn in positive and each column in negative, standard deviation. Output result will give comparison of each column. |
Usage |
col_sig -i file1 -j file2 >out |
-i |
Input file1 of positive example |
-j |
Input file2 of negative example |
file1 |
# Amino Acid Composition of proteins# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y7.88,1.27,4.05,6.39,3.62,7.88,2.13,6.61,5.75,10.23,3.62,2.98,3.41,5.11,5.54,6.39,4.47,7.03,1.70,3.835.46,1.12,7.14,6.72,3.78,5.46,1.68,4.76,6.58,10.64,0.98,7.42,2.52,4.06,4.90,9.38,8.54,5.04,0.98,2.808.96,2.06,4.82,7.58,4.13,6.20,0.69,4.82,7.58,13.10,1.37,2.75,4.82,8.96,6.89,4.13,2.75,6.20,0.00,2.64 |
file2 |
# Amino Acid Composition of proteins# A , C , D , E , F , G , H , I , K , L , M , N , P , Q , R , S , T , V , W , Y8.55,0.65,3.94,9.86,2.63,8.55,0.00,3.28,11.84,13.81,2.63,3.28,1.31,3.94,3.94,6.57,1.31,8.55,0.65,1.8913.29,2.53,3.16,2.53,5.69,14.55,1.89,5.69,3.16,6.32,3.16,3.16,6.96,2.53,6.32,6.32,2.53,6.32,3.16,2.189.88,0.48,7.45,8.91,5.18,3.56,0.48,4.70,11.02,9.88,2.10,6.48,3.89,4.21,3.24,5.99,5.02,2.91,0.16,4.87 |
out |
# Parameters Measured: % Difference, Significance, Average1, Average2, Standard Deviation(SD1), SD2Column 1: -34.83, -490.31, 7.43, 10.57, 0.88, 0.39Column 2: 19.44, 69.33, 1.48, 1.22, 0.33, 0.42Column 3: 9.51, 53.98, 5.34, 4.85, 0.29, 1.50Column 4: -2.89, -28.15, 6.90, 7.10, 0.39, 1.04Column 5: -15.71, -234.15, 3.84, 4.50, 0.16, 0.39Column 6: -30.79, -145.77, 6.51, 8.89, 0.18, 3.07Column 7: 61.49, 218.36, 1.50, 0.79, 0.46, 0.17Column 8: 16.83, 408.83, 5.4, 4.56, 0.33, 0.07Column 9: -26.55, -214.22, 6.64, 8.67, 0.54, 1.35Column 10: 12.34, 240.14, 11.32, 10.01, 1.02, 0.07Column 11: -27.64, -193.90, 1.99, 2.63, 0.35, 0.30Column 12: 1.76, 6.98, 4.38, 4.31, 0.94, 1.25Column 13: -12.27, -115.47, 3.58, 4.05, 0.71, 0.09Column 14: 51.68, 241.21, 6.04, 3.56, 1.68, 0.37Column 15: 24.79, 185.57, 5.78, 4.50, 0.64, 0.73Column 16: 5.22, 41.72, 6.63, 6.30, 1.44, 0.17Column 17: 56.04, 174.63, 5.26, 2.95, 1.44, 1.19Column 18: 2.69, 17.94, 6.09, 5.93, 0.06, 1.74Column 19: -38.94, -72.75, 0.89, 1.32, 0.516, 0.67Column 20: 3.47, 15.63, 3.093, 2.98, 0.26, 1.09 |
Title |
Description |
|
|
seq2pssm_imp (To calculate PSSM matrix in column format without any normalization)The PSSM for each sequence was generated by performing PSI-BLAST search against specific database (e.g. nr) using different iterations (e.g. 3) with cut off e-value 0.001. For a sequence of length N residues, PSSM is represented by an NX20 matrix. Each element of this matrix, m [i, j], provides information on evolutionary conservation of residue type j at sequence position i. For example: |
Usage |
seq2pssm_imp -i seq1.fa -o pssm.out –d nr |
-i |
Input file in the fasta format (not use single fasta format) |
-o |
Output file |
-d |
Database against which PSSM profile is generated |
seq1.fa |
>1BISA PDBID CHAIN_SEQUENCGSHMHGQVDCSPGIWQLDCTHLEGKVILVAVHVASGYIEAEVIPAETGQETAYFLLKLAGRWPVKTVHTDNGSNFTSTTVKAACEWAGIKQEFGIPYNPQSQGVIESMNKELK |
pssm.out |
G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300S, 100, -100, 0, 0, -200, 0, -100, -200, 0, -200, -100, 100, -100, 0, -100, 400, 100, -200, -300, -200H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200, -300, -200, 200M, -100, -100, -300, -200, 0, -300, -200, 100, -100, 200, 499, -200, -200, 0, -100, -100, -100, 100, -100, -100H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200,\-300, -200, 200G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300Q, -100, -300, 0, 200, -300, -200, 0, -300, 100, -200, 0, 0, -100, 499, 100, 0, -100, -200, -200, -100………. |

Title |
Description |
|
|
pssm_n1 (To normalize pssm profile based on 1/(1+e-x) formula)The value of PSSM matrix varies between large range which make difficult for SVM training. Thus every element of PSSM is normalized by using 1/(1+e-x) for normalization.Various formulae can be used for normalization. |
Usage |
pssm_n1 -i pssm.out –o pssm_n1 |
-i |
Input file having pssm profile generated by using (seq2pssm_imp.pl -i seq1.fa -opssm.out –d nr.02) |
-o |
Output file having normalized value |
pssm.out |
G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300S, 100, -100, 0, 0, -200, 0, -100, -200, 0, -200, -100, 100, -100, 0, -100, 400, 100, -200, -300, -200H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200, -300, -200, 200M, -100, -100, -300, -200, 0, -300, -200, 100, -100, 200, 499, -200, -200, 0, -100, -100, -100, 100, -100, -100H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200,-300, -200, 200G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300Q, -100, -300, 0, 200, -300, -200, 0, -300, 100, -200, 0, 0, -100, 499, 100, 0, -100, -200, -200, -100………. |
pssm_n1 |
G, 0.5, 5.19e-131, 3.73e-44, 1.39e-87, 5.19e-131, 1, 1.39e-87, 1.93e-174, 1.39e-87, 1.93e-174, 5.19e-131, 0.5, 1.39e-87, 1.39e-87, 1.39e-87, 0.5, 1.39e-87, 5.19e-131, 1.39e-87, 5.19e-131S, 1, 3.73e-44, 0.5, 0.5, 1.39e-87, 0.5, 3.73e-44, 1.39e-87, 0.5, 1.39e-87, 3.73e-44, 1, 3.73e-44, 0.5, 3.73e-44, 1, 1, 1.39e-87, 5.19e-131, 1.39e-87H, 1.39e-87, 5.19e-131, 3.73e-44, 0.5, 3.73e-44, 1.39e-87, 1, 5.19e-131, 3.73e-44, 5.19e-131, 1.39e-87, 1, 1.39e-87, 0.5, 0.5, 3.73e-44, 1.39e-87, 5.19e-131, 1.39e-87, 1M, 3.73e-44, 3.73e-44, 5.19e-131, 1.39e-87, 0.5, 5.19e-131, 1.39e-87, 1, 3.73e-44, 1, 1, 1.39e-87, 1.39e-87, 0.5, 3.73e-44, 3.73e-44, 3.73e-44, 1, 3.73e-44, 3.73e-44H, 1.39e-87, 5.19e-131, 3.73e-44, 0.5, 3.73e-44, 1.39e-87, 1, 5.19e-131, 3.73e-44, 5.19e-131, 1.39e-87, 1, 1.39e-87, 0.5, 0.5, 3.73e-44, 1.39e-87, 5.19e-131, 1.39e-87, 1G, 0.5, 5.19e-131, 3.73e-44, 1.39e-87, 5.19e-131, 1, 1.39e-87, 1.93e-174, 1.39e-87, 1.93e-174, 5.19e-131, 0.5, 1.39e-87, 1.39e-87, 1.39e-87, 0.5, 1.39e-87, 5.19e-131, 1.39e-87, 5.19e-131Q, 3.73e-44, 5.19e-131, 0.5, 1, 5.19e-131, 1.39e-87, 0.5, 5.19e-131, 1, 1.39e-87, 0.5, 0.5, 3.73e-44, 1, 1, 0.5, 3.73e-44, 1.39e-87, 1.39e-87, 3.73e-44 |
Title |
Description |
|
|
pssm_n2 (To normalize pssm profile based on (numb -min)/(max -min) formula)
|
Usage |
pssm_n2 -i pssm.out –o pssm_n2 |
-i |
Input file having pssm profile generated by using (seq2pssm_imp.pl -i seq1.fa -opssm.out –d nr.02) |
-o |
Output file having normalized value |
pssm.out |
G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300S, 100, -100, 0, 0, -200, 0, -100, -200, 0, -200, -100, 100, -100, 0, -100, 400, 100, -200, -300, -200H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200, -300, -200, 200M, -100, -100, -300, -200, 0, -300, -200, 100, -100, 200, 499, -200, -200, 0, -100, -100, -100, 100, -100, -100H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200,-300, -200, 200G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300Q, -100, -300, 0, 200, -300, -200, 0, -300, 100, -200, 0, 0, -100, 499, 100, 0, -100, -200, -200, -100………. |
pssm_n2 |
G, 0.26, 0.06, 0.20, 0.13, 0.06, 0.66, 0.13, 0, 0.13, 0, 0.06, 0.26, 0.13, 0.13, 0.13, 0.26, 0.13, 0.06, 0.13, 0.06S, 0.33, 0.20, 0.26, 0.26, 0.13, 0.26, 0.20, 0.13, 0.26, 0.13, 0.20, 0.33, 0.20, 0.26, 0.20, 0.53, 0.33, 0.13, 0.06, 0.13H, 0.13, 0.06, 0.20, 0.26, 0.20, 0.13, 0.79, 0.06, 0.20, 0.06, 0.13, 0.33, 0.13, 0.26, 0.26, 0.20, 0.13, 0.06, 0.13, 0.40M, 0.20, 0.20, 0.06, 0.13, 0.26, 0.06, 0.13, 0.33, 0.20, 0.40, 0.59, 0.13, 0.13, 0.26, 0.20, 0.20, 0.20, 0.33, 0.20, 0.20H, 0.13, 0.06, 0.20, 0.26, 0.20, 0.13, 0.79, 0.06, 0.20, 0.06, 0.13, 0.33, 0.13, 0.26, 0.26, 0.20, 0.13, 0.06, 0.13, 0.40G, 0.26, 0.06, 0.20, 0.13, 0.06, 0.66, 0.13, 0, 0.13, 0, 0.06, 0.26, 0.13, 0.13, 0.13, 0.26, 0.13, 0.06, 0.13, 0.06Q, 0.20, 0.06, 0.26, 0.40, 0.06, 0.13, 0.26, 0.06, 0.33, 0.13, 0.26, 0.26, 0.20, 0.59, 0.33, 0.26, 0.20, 0.13, 0.13, 0.2 |

Title |
Description |
|
|
pssm_n3 (To normalize pssm profile based on (numb -min)*100/(max -min) formula)The value of PSSM matrix varies between large ranges which make difficult for SVM training. Thus every element of PSSM is normalized by using (numb -min)*100/(max -min)for normalization. |
Usage |
pssm_n3 -i pssm.out –o pssm_n3 |
-i |
Input file having pssm profile generated by using (seq2pssm_imp.pl -i seq1.fa -o pssm.out –d nr.02) |
-o |
Output file having normalized value |
pssm.out |
G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300S, 100, -100, 0, 0, -200, 0, -100, -200, 0, -200, -100, 100, -100, 0, -100, 400, 100, -200, -300, -200H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200, -300, -200, 200M, -100, -100, -300, -200, 0, -300, -200, 100, -100, 200, 499, -200, -200, 0, -100, -100, -100, 100, -100, -100H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200,-300, -200, 200G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300Q, -100, -300, 0, 200, -300, -200, 0, -300, 100, -200, 0, 0, -100, 499, 100, 0, -100, -200, -200, -100 |
pssm_n3 |
G, 26.68, 6.67, 20.01, 13.34, 6.67, 66.64, 13.34, 0, 13.34, 0, 6.67, 26.68, 13.34, 13.34, 13.34, 26.68, 13.34, 6.67, 13.34, 6.67S, 33.35, 20.01, 26.68, 26.68, 13.34, 26.68, 20.01, 13.34, 26.68, 13.34, 20.01, 33.35, 20.01, 26.68, 20.01, 53.36, 33.35, 13.34, 6.67, 13.34H, 13.34, 6.67, 20.01, 26.68, 20.01, 13.34, 79.98, 6.67, 20.01, 6.67, 13.34, 33.35, 13.34, 26.68, 26.68, 20.01, 13.34, 6.67, 13.34, 40.02M, 20.01, 20.01, 6.67, 13.34, 26.68, 6.67, 13.34, 33.35, 20.01, 40.02, 59.97, 13.34, 13.3, 26.68, 20.01, 20.01, 20.01, 33.35, 20.01, 20.01H, 13.34, 6.67, 20.01, 26.68, 20.01, 13.34, 79.98, 6.67, 20.01, 6.67, 13.34, 33.35, 13.34, 26.68, 26.68, 20.01, 13.34, 6.67, 13.34, 40.02G, 26.68, 6.67, 20.01, 13.34, 6.67, 66.64, 13.34, 0, 13.34, 0, 6.67, 26.68, 13.34, 13.34, 13.34, 26.68, 13.34, 6.67, 13.34, 6.67Q, 20.01, 6.67, 26.68, 40.02, 6.67, 13.34, 26.68, 6.67, 33.35, 13.34, 26.68, 26.68, 20.01, 59.97, 33.35, 26.68, 20.01, 13.34 |
Title |
Description |
|
|
pssm_n4 (To normalize pssm profile based on 1/(1+e-(x/100) formula)The value of PSSM matrix varies between large range which make difficult for SVM training. Thus every element of PSSM is normalized by using 1/(1+e-(x/100) for normalization. |
Usage |
pssm_n4 -i pssm.out –o pssm_n4 |
-i |
Input file having pssm profile generated by using (seq2pssm_imp.pl -i seq1.fa -opssm.out –d nr.02) |
-o |
Output file having normalized value |
pssm.out |
G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300S, 100, -100, 0, 0, -200, 0, -100, -200, 0, -200, -100, 100, -100, 0, -100, 400, 100, -200, -300, -200H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200, -300, -200, 200M, -100, -100, -300, -200, 0, -300, -200, 100, -100, 200, 499, -200, -200, 0, -100, -100, -100, 100, -100, -100H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200,-300, -200, 200G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300Q, -100, -300, 0, 200, -300, -200, 0, -300, 100, -200, 0, 0, -100, 499, 100, 0, -100, -200, -200, -100………. |
pssm_n4 |
G, 0.5, 0.04, 0.26, 0.11, 0.04, 0.99, 0.11, 0.01, 0.11, 0.01, 0.0474258731775668, 0.5, 0.11, 0.11, 0.11, 0.5, 0.11, 0.047, 0.11, 0.04S, 0.73, 0.26, 0.5, 0.5, 0.11, 0.5, 0.26, 0.11, 0.5, 0.11, 0.26, 0.73, 0.26, 0.5, 0.26, 0.98, 0.73, 0.11, 0.04, 0.11H, 0.11, 0.04, 0.26, 0.5, 0.26, 0.11, 0.99, 0.04, 0.26, 0.04, 0.11, 0.73, 0.119202922022118, 0.5, 0.5, 0.26, 0.11, 0.04, 0.11, 0.88M, 0.26, 0.26, 0.04, 0.11, 0.5, 0.04, 0.11, 0.73, 0.26, 0.88, 0.99, 0.11, 0.11, 0.5, 0.26, 0.26, 0.26, 0.73, 0.26, 0.26H, 0.11, 0.047, 0.26, 0.5, 0.26, 0.11, 0.99, 0.047, 0.26, 0.04, 0.11, 0.73, 0.11, 0.5, 0.5, 0.26, 0.11, 0.04, 0.11, 0.88G, 0.5, 0.04, 0.26, 0.11, 0.04, 0.99, 0.11, 0.01, 0.11, 0.01, 0.04, 0.5, 0.11, 0.11, 0.11, 0.5, 0.11, 0.04, 0.11, 0.04Q, 0.26, 0.04, 0.5, 0.88, 0.04, 0.11, 0.5, 0.04, 0.73, 0.11, 0.5, 0.5, 0.26, 0.99, 0.73, 0.5, 0.26, 0.11, 0.11, 0.26 |
Title |
Description |
|
|
pssm_comp (To compute PSSM composition (400 points))Here pssm matrix is coverted in a vector of dimension 400, by computing composition of occurrences of each type of amino acid corresponding to each type of amino acids in protein sequence. It means for each column we will have 20 values instead of one. Every element in this input vector was subsequently divided by the length of the sequence. The resultant matrix with 400 elements was used as input feature for SVM. |
Usage |
pssm_comp -i pssm_n4 –o pssm_n4.out |
-i |
Input file having pssm profile generated by using (seq2pssm_imp -i seq1.fa –o pssm.out –d nr.02) and then scaled by using (pssm_n4.pl -i pssm.out –o pssm_n4) |
-o |
Output file having 400 elements |
pssm_n4 |
G, 0.5, 0.04, 0.26, 0.11, 0.04, 0.99, 0.11, 0.01, 0.11, 0.01, 0.04, 0.5, 0.11, 0.11, 0.11, 0.5, 0.11, 0.04, 0.11, 0.04S, 0.73, 0.26, 0.5, 0.5, 0.11, 0.5, 0.26, 0.11, 0.5, 0.11, 0.26, 0.73, 0.26, 0.5, 0.26, 0.98, 0.73, 0.11, 0.04, 0.11H, 0.11, 0.04, 0.26, 0.5, 0.26, 0.11, 0.99, 0.047, 0.26, 0.047, 0.11, 0.73, 0.11, 0.5, 0.5, 0.26, 0.11, 0.04, 0.11, 0.88M, 0.26, 0.26, 0.04, 0.11, 0.5, 0.04, 0.11, 0.73, 0.26, 0.88, 0.99, 0.11, 0.11, 0.5, 0.26, 0.26, 0.26, 0.73, 0.26, 0.26H, 0.11, 0.04, 0.26, 0.5, 0.26, 0.11, 0.99, 0.04, 0.26, 0.04, 0.11, 0.73, 0.11, 0.5, 0.5, 0.26, 0.11, 0.04, 0.11, 0.88G, 0.5, 0.04, 0.26, 0.11, 0.04, 0.99, 0.11, 0.01, 0.11, 0.01, 0.04, 0.5, 0.11, 0.11, 0.11, 0.5, 0.11, 0.04, 0.11, 0.04Q, 0.26, 0.04, 0.5, 0.88, 0.04, 0.11, 0.5, 0.04, 0.73, 0.11, 0.5, 0.5, 0.26, 0.99, 0.73, 0.5, 0.26, 0.11, 0.11, 0.26 |
pssm_n4.out |
0.98, 0.50, 0.11, 0.26, 0.11, 0.50, 0.11, 0.26, 0.26, 0.26, 0.26, 0.11, 0.26, 0.26894142, 0.26, 0.73, 0.50, 0.50, 0.04, 0.11, 0.50, 0.99, 0.04, 0.01, 0.11, 0.04, 0.04, 0.26, 0.04, 0.26, 0.26, 0.04, 0.04, 0.04, 0.04, 0.26, 0.26, 0.26, 0.11, 0.11, …….. |
Vector |
400 |
Title |
Description |
|
|
pssm2pat (To generate patterns of given size from PSSM matrix)Here we generate PSSM matrix from different window size. If we want to generate 5-window matrix, take two nucleotide matrix forms upstream and downstream and concatenates all matrix in sequential order. For example: pattern matrix of GSHMH, add matrix of each nucleotide it makes the 100 vector long matrix representing H (middle) of nucleotide. For starting nucleotide zero (0) is considered two upstream nucleotide. |
Usage |
pssm2pat -i pssm.out –o pssm_pat –w 5 |
-i |
Input file having pssm profile generated by using (seq2pssm_imp.pl -i seq1.fa -o pssm.out –d nr.02) |
-o |
Output file |
-w |
Window size generated from PSSM matrix |
pssm.out |
G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300S, 100, -100, 0, 0, -200, 0, -100, -200, 0, -200, -100, 100, -100, 0, -100, 400, 100, -200, -300, -200H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200, -300, -200, 200M, -100, -100, -300, -200, 0, -300, -200, 100, -100, 200, 499, -200, -200, 0, -100, -100, -100, 100, -100, -100H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200, -300, -200, 200G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300Q, -100, -300, 0, 200, -300, -200, 0, -300, 100, -200, 0, 0, -100, 499, 100, 0, -100, -200, -200, -100 |
pssm_pat |
# Pattern Window size 5 generated from PSSM matrix. Each line represents pattern for central residue0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,-300,-100,-200,-300,599,-200,-400,-200,-400,-300,0,-200,-200,-200,0,-200,-300,-200,-300,100,-100,0,0,-200,0,-100,-200,0,-200,-100,100,-100,0,-100,400,100,-200,-300,-200,-200,-300,-100,0,-100,-200,799,-300,-100,-300,-200,100,-200,0,0,-100,-200,-300,-200,200 0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,-300,-100,-200,-300,599,-200,-400,-200,-400,-300,0,-200,-200,-200,0,-200,-300,-200,-300,100,-100,0,0,-200,0,-100,-200,0,-200,-100,100,-100,0,-100,400,100,-200,-300,-200,-200,-300,-100,0,-100,-200,799,-300,-100,-300,-200,100,-200,0,0,-100,-200,-300,-200,200,-100,-100,-300,-200,0,-300,-200,100,-100,200,499,-200,-200,0,-100,-100,-100,100,-100,-100………………………………………………… |
Vector |
20*window size (20*5=100) |
Title |
Description |
|
|
pssm_smooth (To designed smooth pssm profile for plot)Here we generate smooth matrix from different window size. If we want to generate 5-window matrix, take two nucleotide matrix forms upstream and downstream and add all five values from each column and divided by five to make average. The matrix of each nucleotide is 20. Each matrix represents about it matrix neighbour nucleotide. Therefore a smooth graph will be generated. |
Usage |
pssm_smooth -i pssm.out –o pssm_pat –w 5 |
-i |
Input file having pssm profile generated by using (seq2pssm_imp.pl -i seq1.fa -o pssm.out –d nr.02) |
-o |
Output file |
-w |
Window size generated from PSSM matrix |
pssm.out |
G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300S, 100, -100, 0, 0, -200, 0, -100, -200, 0, -200, -100, 100, -100, 0, -100, 400, 100, -200, -300, -200H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200, -300, -200, 200M, -100, -100, -300, -200, 0, -300, -200, 100, -100, 200, 499, -200, -200, 0, -100, -100, -100, 100, -100, -100H, -200, -300, -100, 0, -100, -200, 799, -300, -100, -300, -200, 100, -200, 0, 0, -100, -200, -300, -200, 200G, 0, -300, -100, -200, -300, 599, -200, -400, -200, -400, -300, 0, -200, -200, -200, 0, -200, -300, -200, -300Q, -100, -300, 0, 200, -300, -200, 0, -300, 100, -200, 0, 0, -100, 499, 100, 0, -100, -200, -200, -100 |
smooth.out |
G, -40, -340, -160, -200, -300, 379.2, -60.2, -400, -200, -380, -200.2, 0, -260, -160, -200, 40, -200, -320, -280, -260S, -80, -340, -160, -160, -260, 219.4, 139.6, -380, -180, -360, -180.2, 20, -260, -120, -160, 20, -200, -320, -280, -160H, -80, -340, -160, -160, -260, 219.4, 139.6, -380, -180, -360, -180.2, 20, -260, -120, -160, 20, -200, -320, -280, -160M, -100, -340, -140, -80, -260, 59.6, 179.6, -360, -120, -320, -120.2, 20, -240, 19.8, -100, 20, -180, -300, -280, -120H, -120, -340, -120, 0, -260, -100.2, 219.6, -340, -60, -280, -60.2, 20, -220, 159.6, -40, 20, -160, -280, -280, -80G, -160, -380, -120, 40, -280, -140.2, 239.6, -360, -40, -280, -40.2, 0, -220, 259.4, 0, -60, -200, -280, -260, -60Q, -140, -380, -100, 80, -320, -140.2, 79.8, -360, 0, -260, -0.2, -20, -200, 359.2, 20, -40, -180, -260, -260, -120 |
Vector |
20 |
Title |
Description |
|
|
seq2motif (To create motifs by sliding window of user defined length)This program creates motif of defined length. Optional ‘X’ at the end of sequence is added to make complete pattern. The binary pattern is generated using the motifs |
Usage |
seq2motif -i seq1.fa -o motif.out -w 5 -x y |
-i |
Input file in single fasta format |
-o |
Output file |
-w |
Window size to create a pattern |
seq1.fa |
>seq_1##GSHMHGQVDCSPGIWQLDCTHLEGK |
motif_1.out |
>seq_1XXGSHXGSHMGSHMHSHMHGHMHGQMHGQVHGQVDGQVDCQVDCSVDCSPDCSPGCSPGISPGIWPGIWQGIWQLIWQLDWQLDCQLDCTLDCTHDCTHLCTHLETHLEGHLEGKLEGKXEGKXX |
Title |
Description |
|
|
motif2bin (To make binary input from the multifasta motif file)It generates binary input from the multifasta motif file of fixed length into column format. |
Usage |
motif2bin -i motif_1.out -o bin.out -x y |
-i |
Input in multifasta file (seq2motif.pl -i seq1.fa -o motif.out -w 5) |
-o |
Output file |
-x |
If additional X is added in pattern then y (for yes), or n (for no) |
motif_1.out |
>seq_1XXGSHXGSHMGSHMHSHMHGHMHGQMHGQVHGQVDGQVDCQVDCSVDCSPDCSPGCSPGISPGIWPGIWQGIWQLIWQLDWQLDCQLDCTLDCTHDCTHLCTHLETHLEGHLEGKLEGKXEGKXX |
bin.out |
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 00, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,0, 0, 0, 0, 0,0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0………………………………………… |
Vector |
20*window size (20*5=100) |
Title |
Description |
|
|
blast similarity (To perform blast)Basic Local Alignment Search Tool, or BLAST, is an algorithm for comparing primary biological sequence information, such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences. A BLAST search enables a researcher to compare a query sequence with a library or database of sequences, and identify library sequences that resemble the query sequence above a certain threshold. For example, following the discovery of a previously unknown protein in the mouse, a scientist will typically perform a BLAST search of the protein database (nr) to see if humans carry a similar protein; BLAST will identify sequences in the previously known database that resemble the mouse protein based on similarity of sequence. |
Usage |
blast_similarity -i fasta -d nr -j 3 -e 1 –o blast.out |
-i |
Input file of Fasta format |
-o |
Output result |
-d |
Database used blast |
-j |
Number of Iteration |
-e |
Cut off Evalue |
fasta |
>amla_1ASDATAYAACVAYANMANNNAMAKLAWQAPTCAGYAAKTGCVQRATRQOPKALVNAASDREW>amla_2ACDEFGHIKLMNPQRSTVWMRNRGFGRRELLVAMAMLVSVTGCARHASGARPASTTLPAGADLADRFAELERRYDARLG |
blast.out |
>amla_1 Zero Zero>amla_2 ref|NP_216584.1| blaC [Mycobacterium tuberculosis H37Rv] >gi|158... 2e-27 |