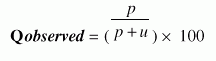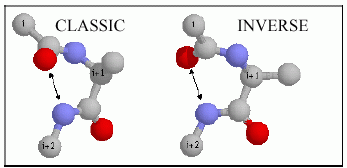
The architecture of protein three-dimensional structure is characterized by the repetitive motif elements such as alpha-helices and beta-sheets, and non-repetitive motif elements such as tight turns, bulges and random coil structures. Tight turns are the third "classical" secondary structures that serve to reverse the direction of the polypeptide chain and are responsible for the globular shape of proteins. They are located primarily on the protein surface and accordingly contain polar and charged residues. Antibody recognition, phophorylation, glycosylation, hydroxylation, and intron/exon splicing are found frequently at or adjacent to turns.
Types of Tight Turns
There are different types of tight turns (Chou 2000) depending upon the number of residues forming the turn. These are as follows:

Method Used
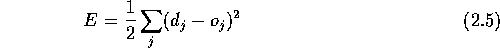
The following figure shows the network architecture used in GammaPred
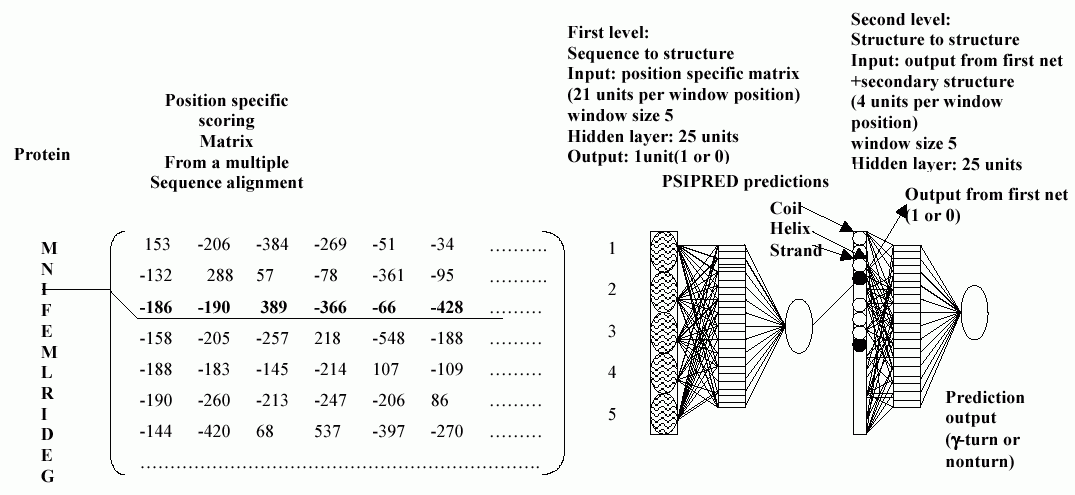
The same approach is used here for the prediction of gamam turns. It is a combination of neural network and multiple alignment information. Net is trained on the PSI-BLAST(part of PSIPRED) generated position specific scoring matrices.
PSI-BLASTThe PSIPRED method has been used for secondary structure prediction. It uses PSI-BLAST to detect distant homologues of a query sequence and generate position specific scoring matrix as part of the prediction process (Jones, 1999), and training is done on these intermediate PSI-BLAST generated position specific scoring matrices as a direct input to the neural network. The matrix has 21 X M elements, where M is the length of the target sequence and each element represents the likelihood of that particular residue substitution at that position in the template. It is a sensistive scoring system, whcih involves the probabilities with which amino acids occur at various positions.
Performance Measures
|

 |
 |