MHC Allele
The major histocompatibility complex (MHC) is a cluster of gene that code for cell
surface proteins which regulate the adaptive immune response. The system is called
H2 in mice and HLA (human lymphocyte antigen) in humans. Class I MHC
contains three genes called HLA-A, B, and C; proteins from these genes are
expressed on almost all cells. Class II MHC genes are called HLA-DR, DQ, and
DP; their proteins are expressed on antigen-presenting macrophages, dendritic
cells and B cells.
The function of these proteins is to present fragments of antigens to T cells. The
receptor of T cells can only recognize antigen fragments in complex with MHC
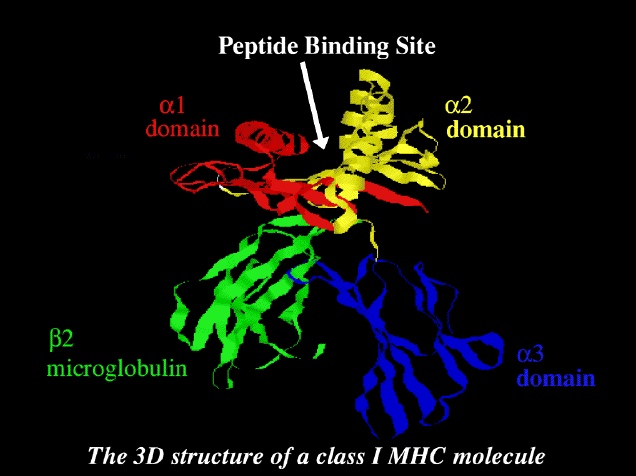
proteins.The detailed structure of MHC class-1 molecule is shown in picture below.
Promiscuous Mutated MHC Binders
An antigen or region of antigenic sequence that after few mutations binds to several MHC alleles.The peptide in its native form binds with a specific MHC allele.These regions are most suitable for vaccine development because
they can generate immune response in large population
Epitope EnhancementThis is an appoarch in which the affinity of peptide toward MHC is increased by modification of peptide.The modification of the peptide mostly obtained through mutating the peptide.This appoarch is known as "Epitope enhancement" due to few observations in literature those shown that MHC binding affinity and immunogenicity of peptide have direct releation.
Single Mutated BindersThe peptide having one amino acid mutated as compared to the native peptide. The length of peptide is always nine. The mutation can be at any position from 1 to 9.The mutation is always subsitution mutation
Double Mutated BindersThe peptide having two amino acid mutated as compared to the native peptide. The length of peptide is always nine. The mutation can be at any two positions from 1 to 9.The value of positions for mutation should be different e.g. 1 & 2, 2 & 3 and so on.
Triple Mutated Binders
The peptide having three amino acid mutated as compared to the native peptide. The length of peptide is always nine. The mutation can be at any three positions from 1 to 9.The value of positions for mutation should be different e.g. 1 ,2 & 3, 5,2 & 9 and so on.
Multiplication MatricesThese are the matrices from which the score of the peptide is obtained by multiplying the score of each position of the peptide.The score of each amino acid at each position are precalculated and stored in 20 X 9 table.
Addition Matrices
These are the matrices from which the score of the peptide is calculated by summing up the the score of each position of the peptide.The score of each amino acid at each position are precalculated and stored in 20 X 9 table.
Threshold Value
A preselected numerical value used to differentiate between binders and non binders. Any peptide
frame scoring higher than this value is predicted as binder or vice versa. The threshold is defined as the 'percentage
of best scoring natural peptides'. For example, a threshold of 1% would predict peptides in any given protein
sequence which belong to the 1% best scoring natural peptides. The threshold correlate with the peptide score (
Sturniolo et al., 1999) and therefore with HLA-ligand interaction. More importantly, threshold is an indicator for the
likelihood that predicted peptide is capable of binding to a given HLA-molecule. The lower the threshold (= high
stringency), the lower the false positive rate and the higher the false negative rate. in contrast the higher the
threshold (= low stringency), the higher the false positive rate and the lower the false negative rate. In short, from the
same protein sequence input, a threshold setting of 1% will predict a lower number of peptide sequences and for a
lower number of HLA-II alleles, compared to 2% or higher thresholds; however, this will ensure a higher likelihood
of positive downstream experimental results. Normally, at least for a first round of screening, threshold values higher
than 3% are not desirable,since the rate of false positives can increase the size of the predicted repertoire to an
amount unacceptable for later experimental testing.
How the threshold value for each allele is derived.
It is important to calculate the threshold score for each allele so that binders and non-binders can be distinguished. Ideally, one needs sufficient number of binders and non-binders to calculate the threshold score. The lack of peptides particularly non-binders make its impossible to calculate the threshold. In order to overcome this problem we have adopted a following uniform procedure for each matrix.
We have obtained the all protein (~88,000) from SWISSPROT databases release 67 and the overlapping peptides of length nine have been generated for all proteins.
For example, a protein of length n will have ( n+1 - 9 ) overlapping peptides.
Score of all natural 9-mer peptides have been calculated using weight matrix of that allele. These peptides have been sorted on the basis of score in descending order and top 1 % natural peptides have been obtained. The minimum score is determined from these selected peptides. This minimum score is called threshold score of at 1% . Similarly, threshold scores at 2%, 3% \'85 10% are calculated.
The step 1 and 2 is repeated for each allele, in order to calculate threshold score at different percent for each allele.